贵州大学李虎,最新Angew!基于榫卯互锁机制的单原子Ba介导氢键相互作用助力纯水中可充电生物质电氧化!王柯平一作
2026-04-15 浏览次数: 10
贵州大学李虎,最新Angew!基于榫卯互锁机制的单原子Ba介导氢键相互作用助力纯水中可充电生物质电氧化!王柯平一作
生物质衍生分子(如5-羟甲基糠醛,HMF)的电氧化升级提供了一种碳中和路线来生产有价值化学品,其转化效率高度依赖于强碱和昂贵膜的使用。
2026年04月13日,贵州大学李虎团队在Angewandte Chemie International Edition期刊发表题为“Single-Atom Barium-Mediated Hydrogen-Bonding Interaction via Mortise-and-Tenon Interlock for Rechargeable Biomass Electrooxidation in Pure Water”的研究论文,团队成员王柯平为论文第一作者,李虎为论文通讯作者

该研究将单原子Ba锚定到羟基氧化镍中作为硫酸根(榫头)的卯眼,构建了一种可充电阳极S-Ba₁Ni₁₋ₓOOH,用于解耦HMF电氧化,从而在无碱、无外加电位和无膜条件下实现2,5-呋喃二甲酸(FDCA)的定量生产。该解耦系统包括:(i) 通过充电实现活性Ni³⁺—O的(再)生成,以及 (ii) 在纯水中无电(放电)条件下,用Ni³⁺—O提取HMF的H原子,通过去质子化生成FDCA(转化率和选择性接近100%)。表面硫酸根与HMF的羟基之间的氢键相互作用可以加速底物向固-液界面的迁移,从而提高转化率。理论计算阐明,氢键相互作用降低了HMF中O—H键的O-2p与H-1s轨道之间的杂化程度,从而提高了其H的脱除能力,进而促进HMF完全氧化为FDCA。多种生物质衍生物也适用于该解耦系统,以>96.3%的产率得到相应的酸。技术经济分析表明,用纯水替代强碱可以将FDCA的生产成本降低50.5%,凸显了该解耦策略的广阔前景,并为生物质高值化提供了一条有吸引力的途径。
生物质高值化提供了极具前景且可持续的路线来生产有价值的化学品,并有望消除对石油基资源的依赖。一个特别引人注目的范例是5-羟甲基糠醛(HMF)氧化为2,5-呋喃二甲酸(FDCA),因为FDCA是石油衍生对苯二甲酸的潜在替代品,后者广泛应用于合成聚合物材料,如聚呋喃二甲酸乙二醇酯、增塑剂和聚酰胺。目前,电催化被认为是一种绿色高效的催化方法来实现该反应,在温和反应条件下可持续生产FDCA,从而避免了热催化的一些局限性,例如需要额外氧化剂以及高温高压。尽管有这些优点,电催化HMF氧化(E-HMFOR)生成FDCA仍然需要碱性条件(通常为1M KOH),此时生成的FDCA在反应后以羧酸盐形式存在于电解液中,需要大量的酸(如H₂SO₄)来中和残余的碱并使FDCA质子化。然而,HMF在碱性条件下容易发生非法拉第副反应,例如低聚和坎尼扎罗反应,这会降低产物的产率和纯度。此外,该转化通常在两室电解槽中进行,因此需要使用阳离子或阴离子交换膜,这无意中增加了操作成本。尽管已有研究表明,较弱的碱性介质可以适当缓解其中一些限制,但仍然面临产物纯化和膜使用的问题。在此背景下,深入研究E-HMFOR中HMF与电极之间的相互作用,可能有助于开发高效合成FDCA的催化体系。
迄今为止,串联“电化学-化学”机制被广泛用于描述E-HMFOR过程,该过程涉及两个独立的反应:i) 电化学步骤(例如,Ni²⁺—OH + OH⁻ → Ni³⁺—O + H₂O):在碱性和阳极电位条件下,Ni²⁺—OH发生去质子化形成活性Ni³⁺—O物种;ii) 化学步骤(例如,Ni³⁺—O + HMF → Ni²⁺—OH + FDCA):电化学生成的Ni³⁺—O作为化学氧化剂,能够实现HMF的自发脱氢生成FDCA,并最终回到Ni²⁺—OH。重要的是,电化学和化学步骤都需要持续供应电流,以促进反应物(如OH⁻和HMF)向固-液界面的迁移,从而使得E-HMFOR顺利发生。已有研究阐明,化学步骤可以在非酸性条件下发生,无需施加电位,此时中性电解液(如H₂O)可以提供稳定的介质以避免HMF降解和FDCA去质子化(图S1),这可能允许HMF氧化在无碱、无膜和无电位的条件下进行。即使电化学步骤可以在无膜体系中进行,它仍然是一个依赖碱性和电位(pH > 13)的过程,导致溶剂不兼容。
在有机电合成和全水解中,类似的电化学-化学循环可以解耦为两个独立的反应,其中电化学步骤和化学步骤可以在不同的电解液中进行。这种解耦策略可以预见能够解决E-HMFOR中溶剂不兼容和膜使用的问题,使得电化学步骤和化学步骤在各自友好的条件下(碱性和中性)独立运行。因此,解耦系统所需的催化剂应满足以下标准:i) 快速形成活性相(电化学步骤),ii) 在无电条件下反应物易于从本体电解液向催化界面迁移,iii) 有利的脱氢反应(化学步骤),以及iv) 强氧化性和高可循环性,这可能需要精细的修饰。由于亲核属性,含氧物种可以促进含氢底物在催化界面的吸附,这有望满足上述标准,但前提是要确保含氧物种的稳定存在,特别是在无电条件下。
受这些发现的启发,在此,该研究将高度分散的Ba单原子插入NiOOH中,作为SO₄²⁻(榫头)的卯眼,通过榫卯互锁使其稳定存在于催化界面(表示为S-Ba₁Ni₁₋ₓOOH),从而将HMF电氧化为FDCA的过程解耦。通过这种方法,HMF氧化为FDCA可以在无膜、无碱和无电条件下进行。在无电条件下,SO₄²⁻与Ba²⁺之间的榫卯互锁能够使SO₄²⁻基团稳定存在于S-Ba₁Ni₁₋ₓOOH上,通过氢键相互作用有效连接固-液界面间隙,加速HMF从本体溶液向催化界面的迁移,有利于HMF氧化。理论计算阐明,氢键效应降低了O 2p和H 1s轨道之间的杂化程度以提高H的迁移率,从而促进O—H键断裂,加速HMF氧化为FDCA。该体系实现了接近100%的FDCA产率和选择性。S-Ba₁Ni₁₋ₓOOH在氧化后可通过充电恢复。此外,所开发的解耦系统通过避免碱/酸的使用可以降低FDCA的生产成本,并可推广用于氧化多种生物质衍生物。该研究强调了HMF氧化的解耦策略的重要性和可行性,为生物质高值化和有机氧化的催化体系和催化剂定制提供了新范例。
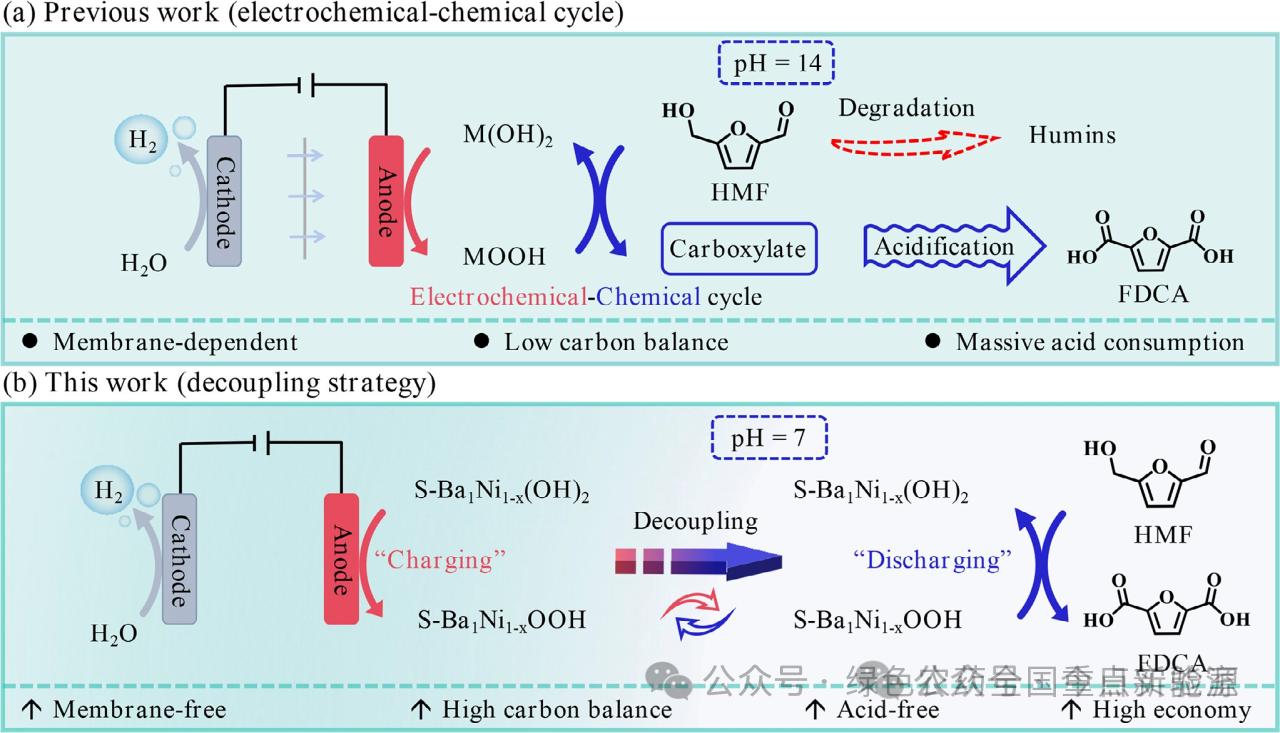
示意图1 | 通过(a)电化学-化学循环(前期工作)和(b)解耦策略(本工作)将HMF电氧化为FDCA的示意图
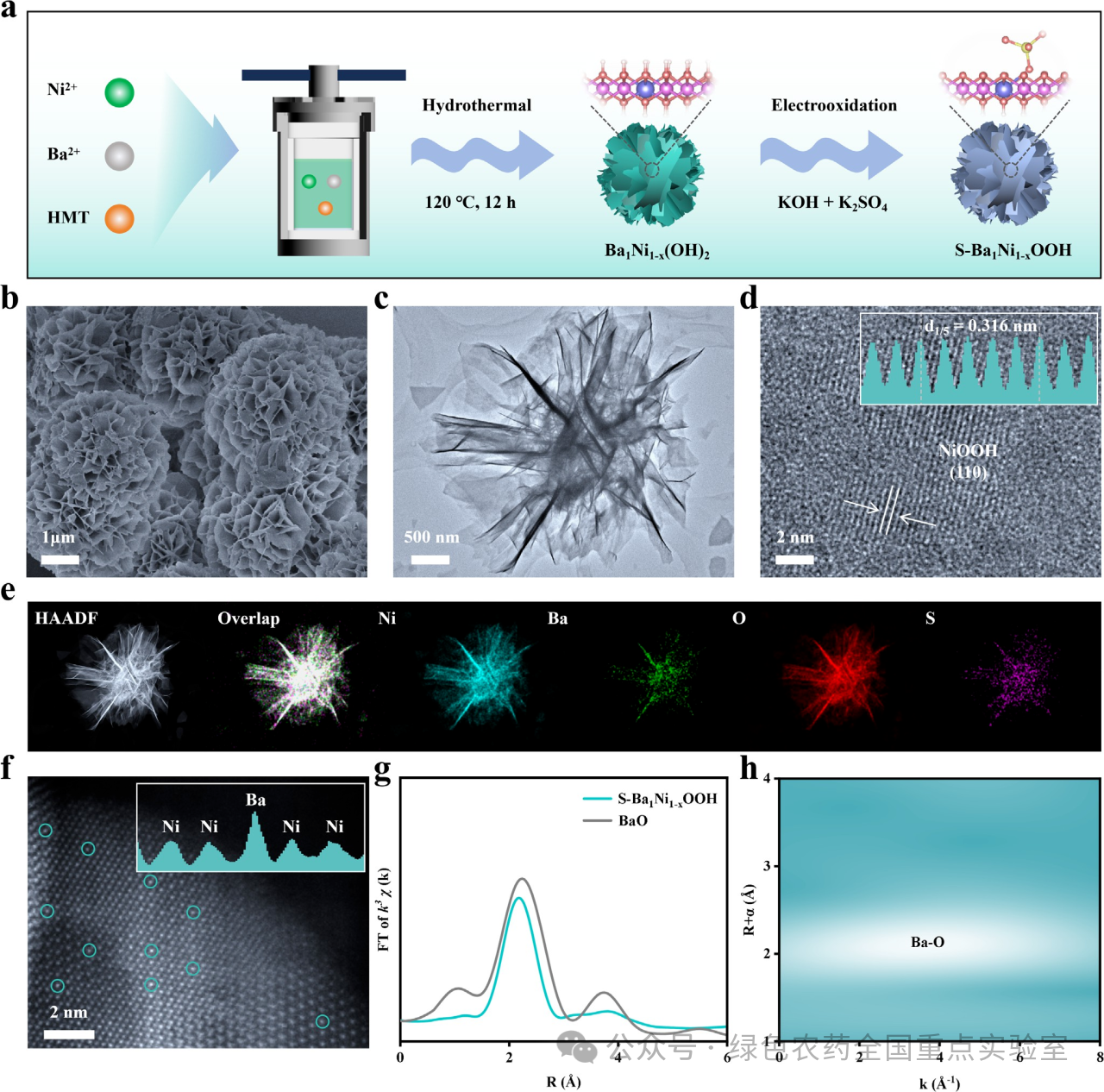
图 1 | 催化剂的合成与表征。(a) 制备S-Ba₁Ni₁₋ₓOOH催化剂的示意图。(b) S-Ba₁Ni₁₋ₓOOH催化剂的SEM图像,(c) TEM图像,(d) 高分辨TEM图像,以及(e) 相应的元素分布图。(f) S-Ba₁Ni₁₋ₓOOH催化剂的AC HAADF-STEM图像(插图:强度分布图)。(g) S-Ba₁Ni₁₋ₓOOH催化剂和BaO参比样品的Ba L边傅里叶变换EXAFS谱图。(h) S-Ba₁Ni₁₋ₓOOH催化剂的小波变换EXAFS信号
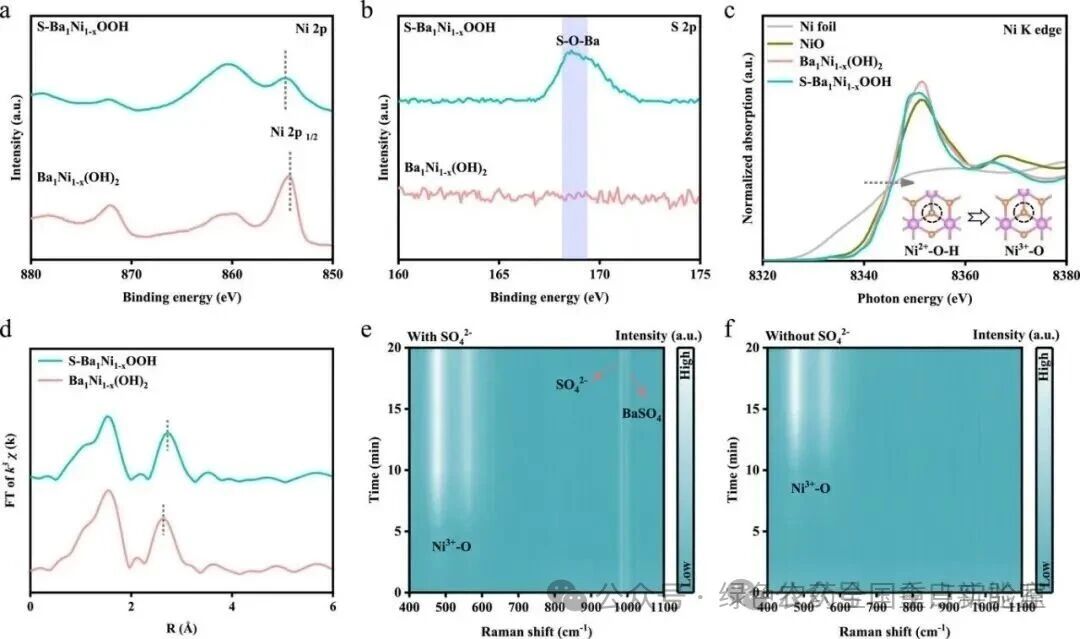
图 2 | 活性相的鉴定。(a) Ba₁Ni₁₋ₓ(OH)₂和S-Ba₁Ni₁₋ₓOOH的Ni 2p XPS谱图和(b) S 2p XPS谱图。(c) Ni箔、NiO、Ba₁Ni₁₋ₓ(OH)₂和S-Ba₁Ni₁₋ₓOOH的Ni K边XANES谱图。(d) Ba₁Ni₁₋ₓ(OH)₂和S-Ba₁Ni₁₋ₓOOH的傅里叶变换EXAFS谱图。在KOH电解液中(e)含有SO₄²⁻和(f)不含SO₄²⁻时,Ba₁Ni₁₋ₓ(OH)₂在1.4 VRHE下的时间依赖拉曼光谱
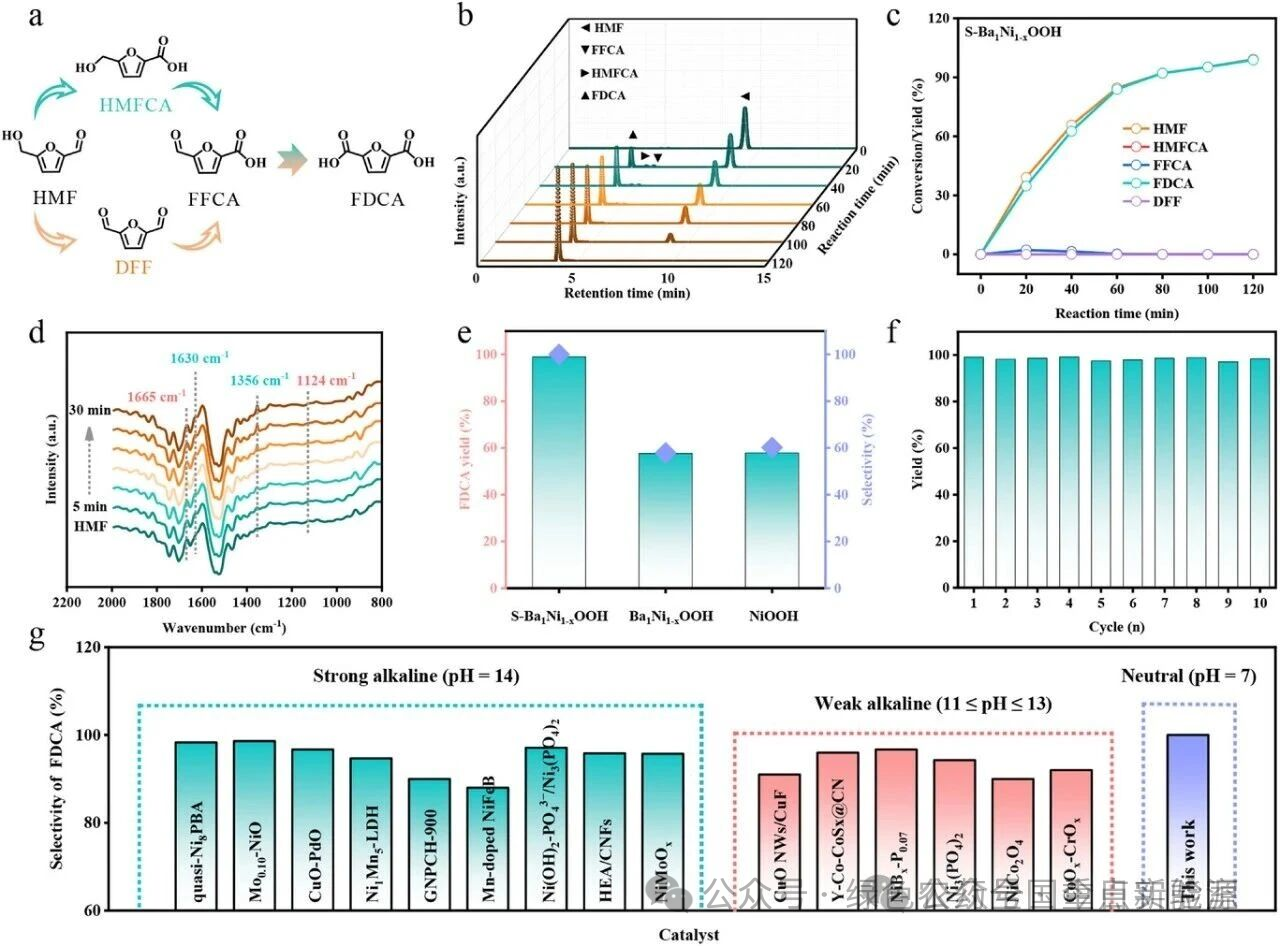
图 3 | HMFOR性能研究。(a) HMF氧化为FDCA的反应路径。(b) S-Ba₁Ni₁₋ₓOOH催化HMF(5 mM, 2 mL)氧化的HPLC谱图。(c) 不同时间下S-Ba₁Ni₁₋ₓOOH催化HMF(5 mM, 2 mL)氧化中FDCA的产率和转化率。(d) S-Ba₁Ni₁₋ₓOOH上HMF氧化的时间依赖原位ATR-IR光谱。(e) 不同催化剂(负载量:6 mg)在HMF(5 mM, 2 mL)氧化120 min后FDCA的产率和选择性。(f) 连续多次HMF氧化中的FDCA产率。(g) S-Ba₁Ni₁₋ₓOOH在HMF氧化中的FDCA选择性与本工作之前报道的催化剂的比较
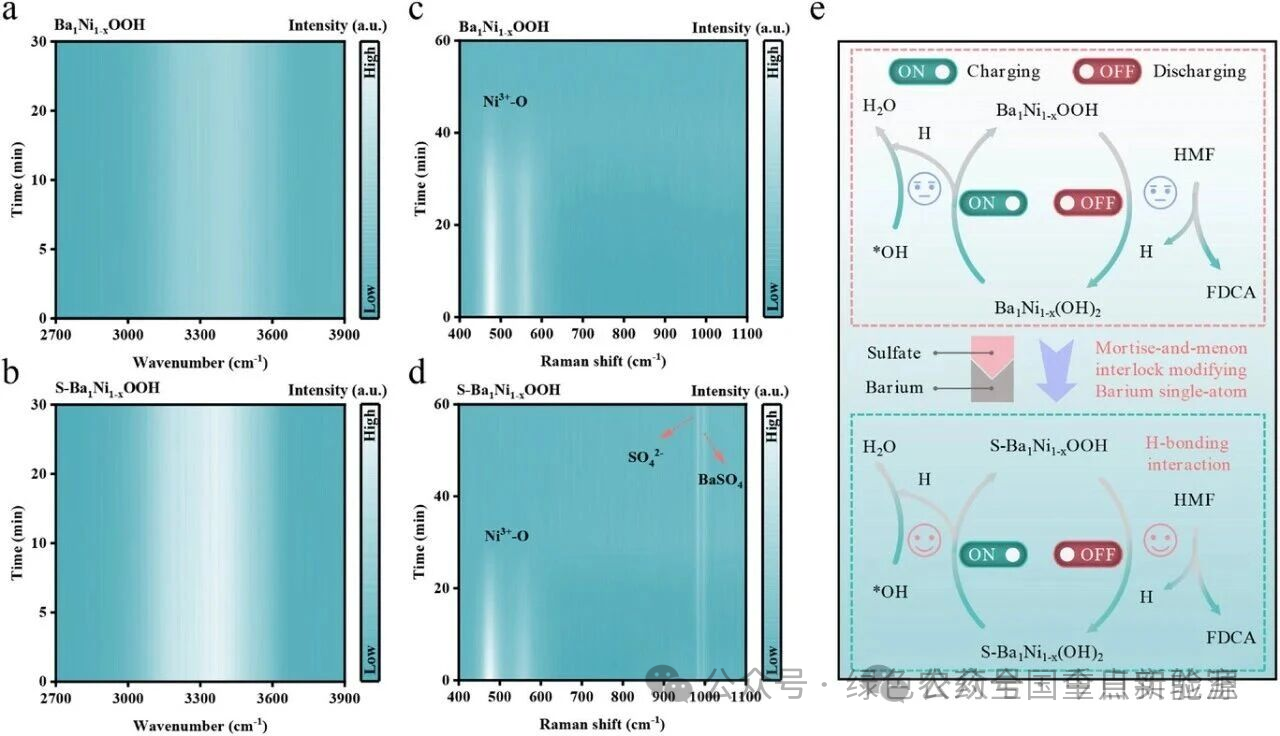
图 4 | HMF氧化的机理研究。使用(a) Ba₁Ni₁₋ₓOOH和(b) S-Ba₁Ni₁₋ₓOOH催化剂在HMF氧化过程中的时间依赖原位ATR-IR光谱。使用(c) Ba₁Ni₁₋ₓOOH和(d) S-Ba₁Ni₁₋ₓOOH催化剂在HMF氧化过程中的时间依赖拉曼光谱。(e) Ba₁Ni₁₋ₓOOH和S-Ba₁Ni₁₋ₓOOH催化剂催化HMF氧化为FDCA的示意图。
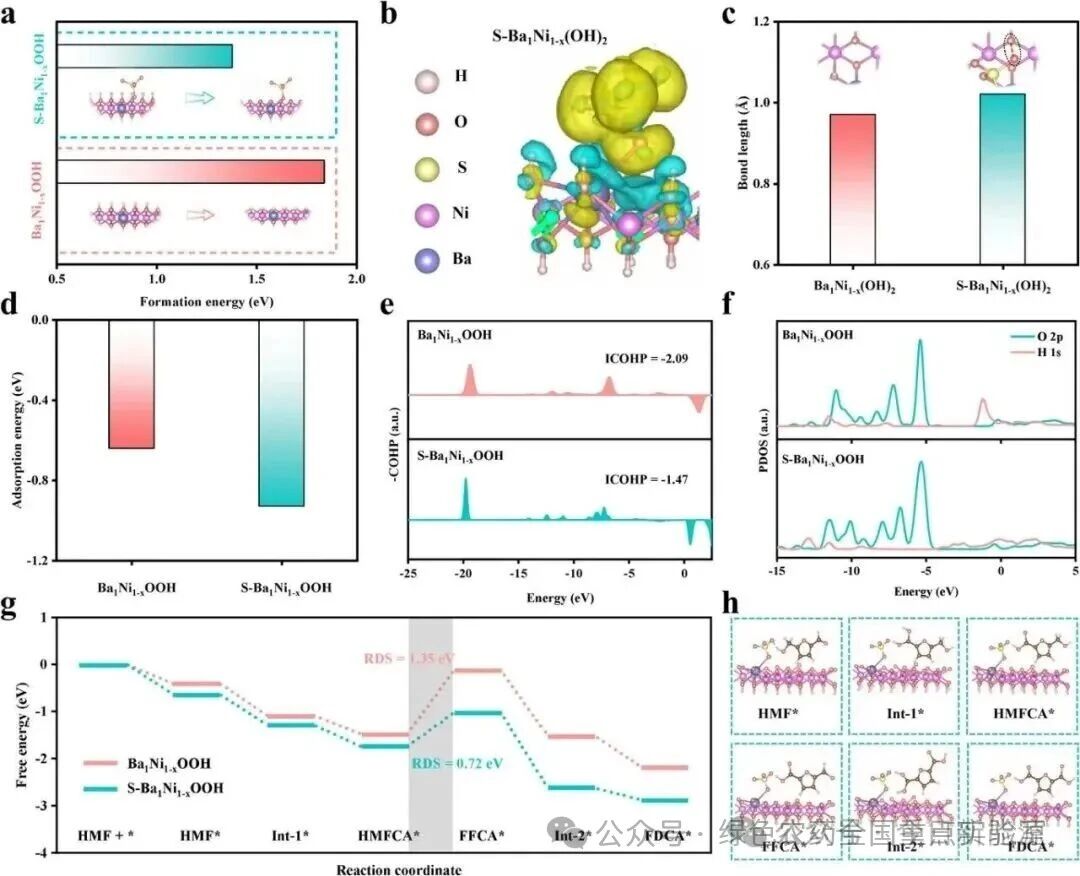
图 5 | DFT计算。(a) S-Ba₁Ni₁₋ₓOOH和Ba₁Ni₁₋ₓOOH的形成能。(b) S-Ba₁Ni₁₋ₓ(OH)₂的电荷密度差图。(c) S-Ba₁Ni₁₋ₓ(OH)₂和Ba₁Ni₁₋ₓ(OH)₂中O—H的键长。(d) HMF在S-Ba₁Ni₁₋ₓOOH和Ba₁Ni₁₋ₓOOH上的吸附能。(e) HMF中O—H的COHP分析和(f) DOS分析。(g) HMF在S-Ba₁Ni₁₋ₓOOH和Ba₁Ni₁₋ₓOOH上氧化为FDCA的吉布斯自由能曲线。(h) HMF在S-Ba₁Ni₁₋ₓOOH上氧化为FDCA的模型演化过程
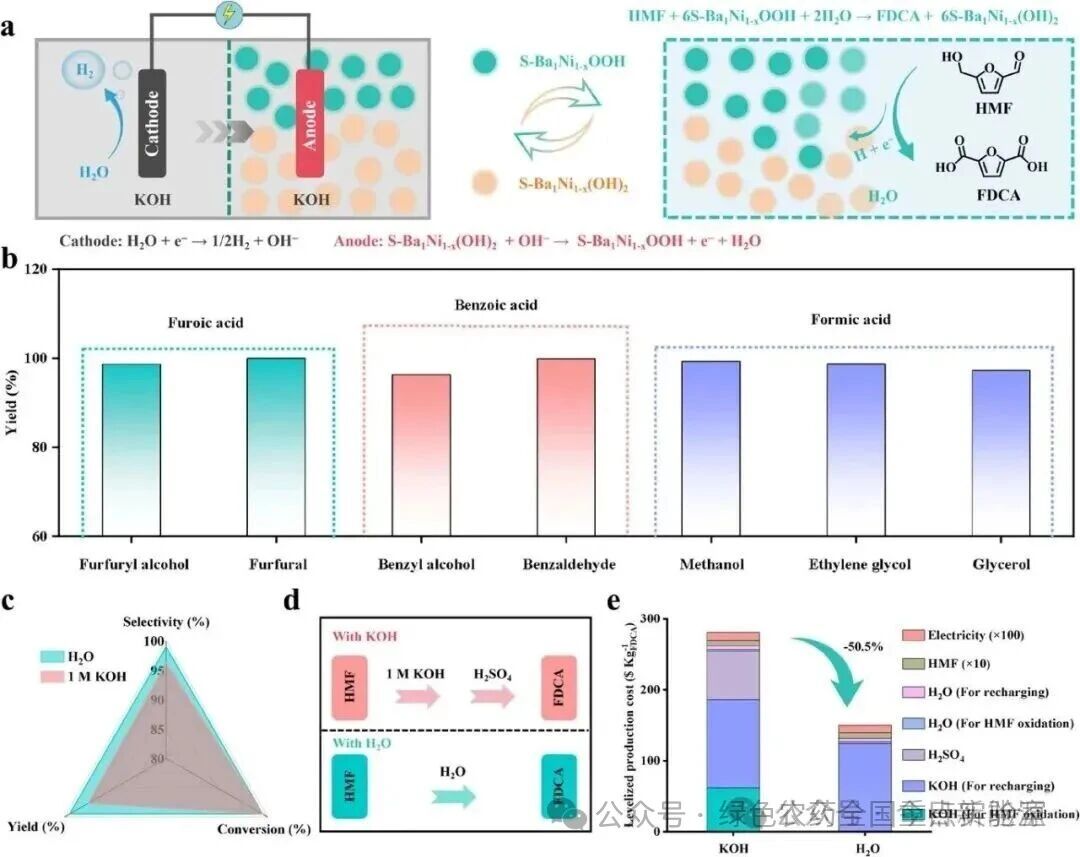
图 6 | 底物范围与技术经济分析。(a) 解耦系统从HMF氧化合成FDCA的示意图。(b) 底物范围拓展实验。(c) 在纯水和1 M KOH中FDCA的选择性、产率和转化率。(d) 在纯水和1 M KOH中从HMF氧化合成FDCA的流程图。(e) FDCA的生产成本。
总之,该研究提出了一种“可充电”S-Ba₁Ni₁₋ₓOOH,用于将HMF电氧化解耦成一个两步过程,从而在无膜、无碱和无外加电位的条件下合成FDCA。利用这种解耦策略,两个不兼容的步骤可以在各自友好的条件下独立运行:i) 通过充电可以(再)生成对碱友好的活性Ni³⁺—O;ii) 在纯水中,对碱敏感的HMF发生去质子化,生成接近100%转化率和选择性的FDCA。结合原位测试和理论计算,证实了S-Ba₁Ni₁₋ₓOOH中硫酸根诱导的氢键相互作用通过促进HMF吸附和O—H去质子化,全面简化了HMF氧化为FDCA的过程,显著将决速步(HMFCA → FFCA)的能垒从1.35 eV降低到0.72 eV。此外,该研究的原料范围可以扩展到多种生物质衍生物,以>96.3%的产率得到相应的酸作为最终产物。该解耦策略用于从HMF氧化生产FDCA具有高度经济性,具有消除对碱/酸和膜依赖的巨大潜力,为产物分离和有机氧化提供了一种有吸引力且可持续的范式。(转载自:今日新能源,2026-04-14)
审校:吴增雪
编审:李向阳
终审:张 林