【转载】【ACS Catal.】贵州大学池永贵:光催化芳基迁移的原位胺化:快速合成4-氨基苯丙酰胺
2026-01-04 浏览次数: 35
【ACS Catal.】贵州大学池永贵:光催化芳基迁移的原位胺化:快速合成4-氨基苯丙酰胺
贵州大学池永贵团队报道了一种基于光催化芳基迁移的烯烃双官能团化方法,可快速获得具有价值的4-氨基苯丙酰胺骨架。该反应通过自由基介导的远程芳烃C-H胺化反应实现4-氨基芳环的原位形成,显著提升了传统芳基迁移策略的灵活性,并提高了4-氨基芳基的迁移效率。该反应适用于多种双功能试剂,可实现含磺酰基、三氟甲基、二氟甲基或氯二氟甲基的苯丙酰胺的发散性合成,收率中等至良好。通过对药用相关分子的后期修饰、产物进一步转化、潜在生物活性分子的合成及抗菌活性研究,验证了该策略的实用价值。
Abstract
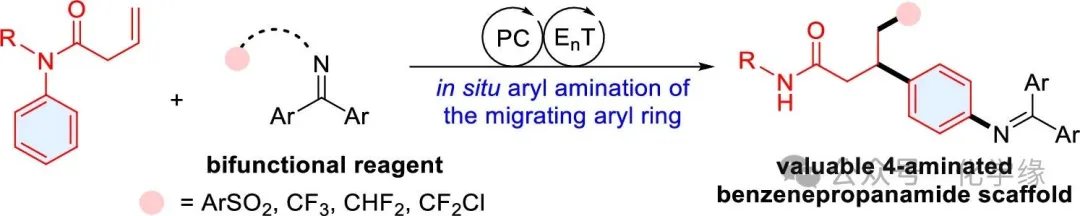
Aryl migration-induced difunctionalization of alkenes is a fascinating strategy for increasing the molecular complexity via the simultaneous formation of two chemical bonds across the C-C double bond. Despite the significant advances in this area, thein situ functionalization of the migrating aryl ring remains elusive due to the incompatibility between the conventional arene C-H functionalization strategy and the aryl migration process. Herein, we disclose the photocatalytic in situ amination of the migrating aryl ring in which an aryl ring is aminated and migrated within a single step, providing rapid access to valuable 4-aminated benzenepropanamide scaffolds. Such transformations enable the formation of an additional chemical bond on the migrating aryl ring beyond those two formed on the alkene carbons, significantly increasing the flexibility of the aryl migration strategy and improving the migration efficiency for the aminated aryl ring. The energy transfer catalytic cycle between the photosensitizer and the bifunctional reagents plays a pivotal role in combining the aryl migration process with the emerging radical-based arene remote C-H amination step. Experimental mechanistic studies support the proposed reaction pathway. The power of this protocol was demonstrated by the functionalization of pharmaceutically relevant molecules, the efficient synthesis of bioactive molecule analogs, and antibacterial activity investigations.
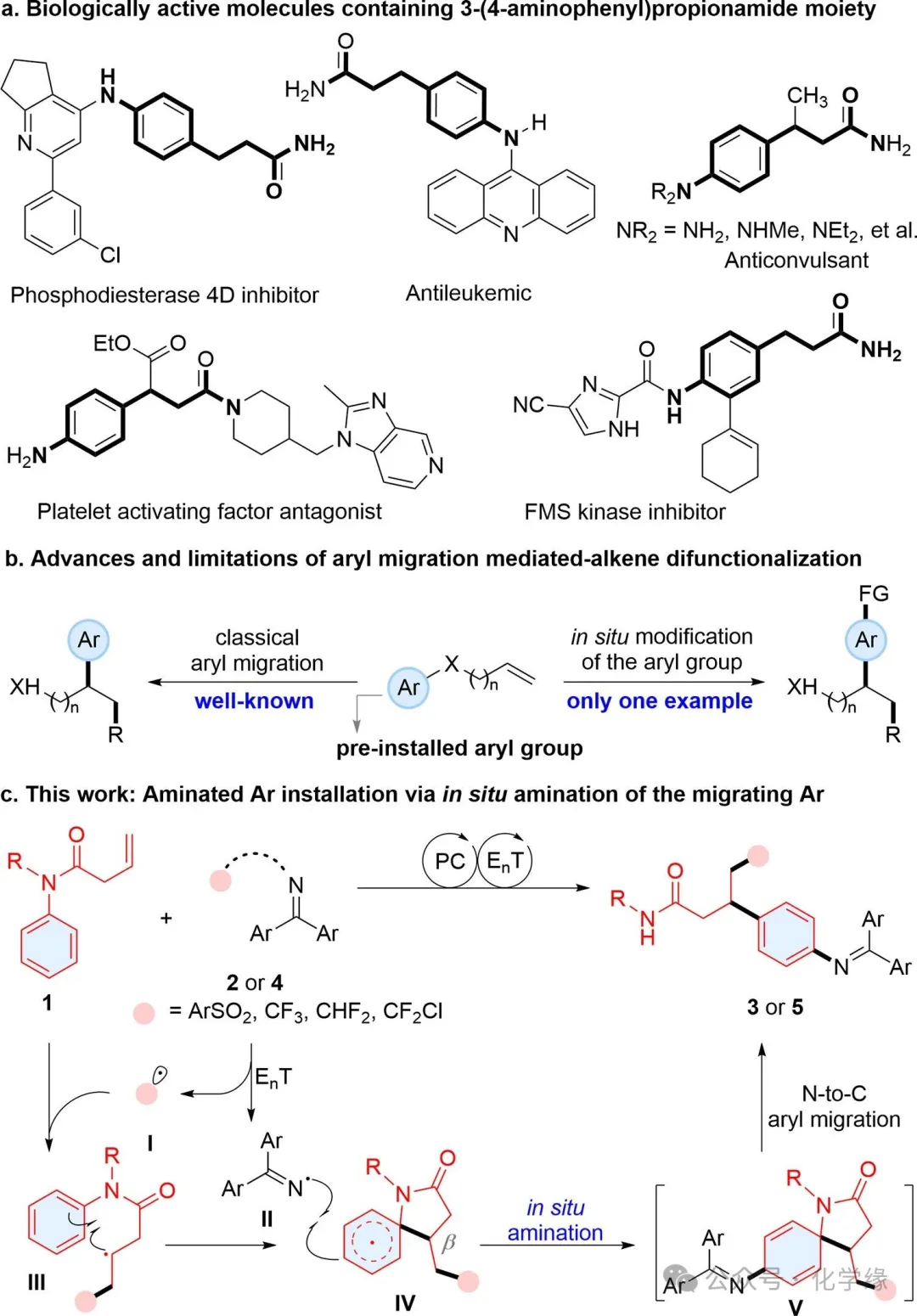
图 1 通过芳基迁移原位胺化反应实现芳环胺化修饰的背景与反应设计
4-氨基苯丙酰胺骨架被设计为核心药效团,展现出抗肿瘤、抗白血病、抗病毒(如登革热病毒和西尼罗河病毒)等活性。因此,开发温和可靠的合成方法以高效构建此类骨架具有重要意义。
芳基迁移介导的烯烃双官能团化是将商品化烯烃快速转化为苯丙酰胺骨架的最简明策略之一。芳基迁移通常始于预制芳基连接烯烃与原位生成的自由基物种反应,继而发生芳基迁移过程,实现预装芳基的迁移。对于某些目标芳基,其预装过程有时合成复杂或难以实现,限制了该策略的普适性。为提升该策略的灵活性并提高特定芳基的迁移效率,研究已取得显著进展。例如,Stephenson团队揭示了烯烃的光催化氨基芳基化反应,通过选择不同芳基磺酰乙酰胺作为双功能试剂,即可高效将各类芳基引入烯烃上。Zhu团队报道了通过新型对接迁移策略实现烯烃的芳基二氟甲基化,其中迁移的芳基参与了双功能试剂的反应。Studer等人披露了一种前所未有的B-C芳基迁移策略,通过原位连接策略实现了迁移芳基的灵活变化。尽管这些开创性策略显著提升了特定芳基团的迁移效率,但双功能试剂的预制要求以及强碱性、亲核性芳基锂试剂的适用性,在一定程度上限制了其应用范围。
芳烃直接C-H官能团化是构建取代芳香化合物的最有效策略,该领域已取得重大进展。然而,在迁移过程中对迁移芳基进行原位官能团化仍难以实现,这可能是由于传统金属催化的芳烃C-H官能团化与自由基驱动的芳基迁移过程存在不相容性。最近,开发了一种概念上全新的自由基介导的芳烃远程C-H官能团化策略,该策略已成功应用于对位选择性芳烃胺化和酰化反应。这种自由基芳烃C-H胺化策略可能与芳基迁移过程相容,为实现迁移芳环的胺化提供了新途径。本文报道了一种光催化芳基迁移策略,通过迁移芳基的原位胺化高效合成4-胺化苯丙酰胺骨架。同时光催化生成两种具有不同反应活性的自由基物种(持久性自由基与瞬态自由基),在将芳基迁移过程与芳烃远程C-H胺化步骤相结合中起着关键作用。瞬态自由基物种(I)与持久性亚胺基自由基(II)分别主导C-C双键的自由基加成反应(步骤I至III)及后续原位胺化过程(步骤IV至V)。惰性C-N键的断裂最终完成4-氨基芳环的迁移,生成具有价值的4-氨基苯丙酰胺骨架。该反应通过迁移芳环的原位胺化,规避了制备对位氨基底物的繁琐合成路线。通过选择不同双功能试剂即可改变所得苯丙酰胺的结构。该反应成功应用于药学相关分子的功能化修饰、生物活性分子类似物的高效分支合成及抗菌活性研究。
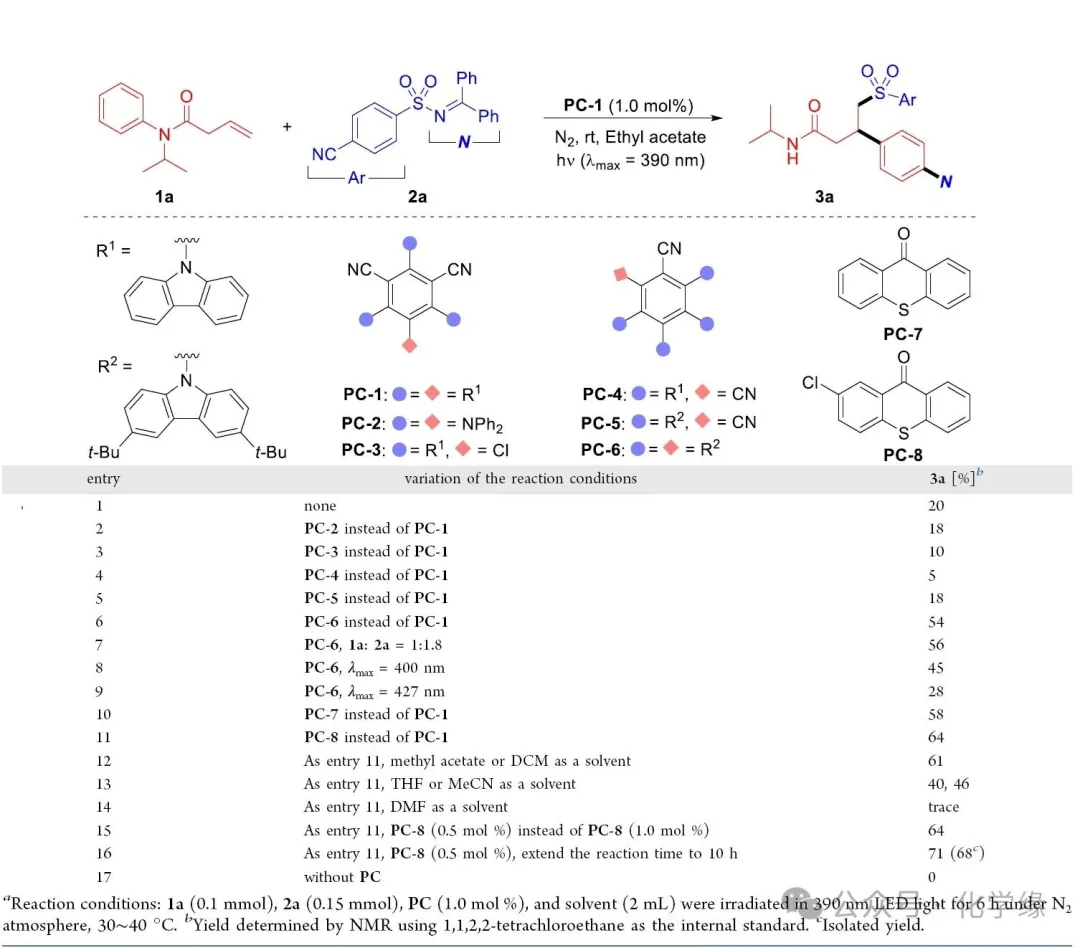
图 2 反应条件优化
Reaction conditions: 1a (0.1 mmol), 2a(0.15 mmol), PC (1.0 mol %), and solvent (2 mL) were irradiated in 390 nm LED light for 6 h under N2 atmosphere, 30∼40 °C.
为验证假说,需选择一种适宜的双功能试剂,使其既能生成瞬态自由基(参与自由基加成步骤I至III),又能形成持久性氮自由基物种(主导自由基重组步骤IV至V)。磺酰亚胺类化合物因能在光催化能量转移(EnT)过程中通过弱N-S键(≈70 kcal/mol)的同解离,同时生成磺酰自由基与亚胺基自由基,已成为强效反应试剂。因此,选取磺酰亚胺2a(1.5 equiv)作为模型双功能试剂,与C-C双键连接的N-苯基酰胺1a(1.0 equiv)反应。初步尝试采用4CzIPN(1 mol%)作为光敏剂,乙酸乙酯为溶剂,在光照(λmax = 390 nm)下反应,得到目标产物苯丙酰胺3a,收率20%。后续对若干氰基苯衍生物光敏剂、的考察未能提高反应收率。五噁唑基氰基苯化合物展现出高催化效率,3a的收率达54%。将2a用量增至1.8当量可略微提高收率。使用更长波长的光源会导致收率下降。鉴于硫氧杂酮(65.5 kcal/mol)的高三重态能及其作为高效能量传递剂的特性,本研究选用噻吨酮PC-7和PC-8催化该转化反应。采用2-氯噻吨酮PC-8作为光敏剂时,3a的收率达64%。溶剂筛选表明:乙酸甲酯与二氯甲烷收率与乙酸乙酯相当;四氢呋喃和乙腈导致收率显著下降;强极性溶剂DMF不适合作为反应溶剂。降低催化剂量(0.5 mol%)仍可维持收率。将反应时间从6小时延长至10小时可进一步提升收率至71%,对应的分离收率为68%。最终对照实验证实光敏剂对反应至关重要:在PC-8缺失时反应完全抑制。
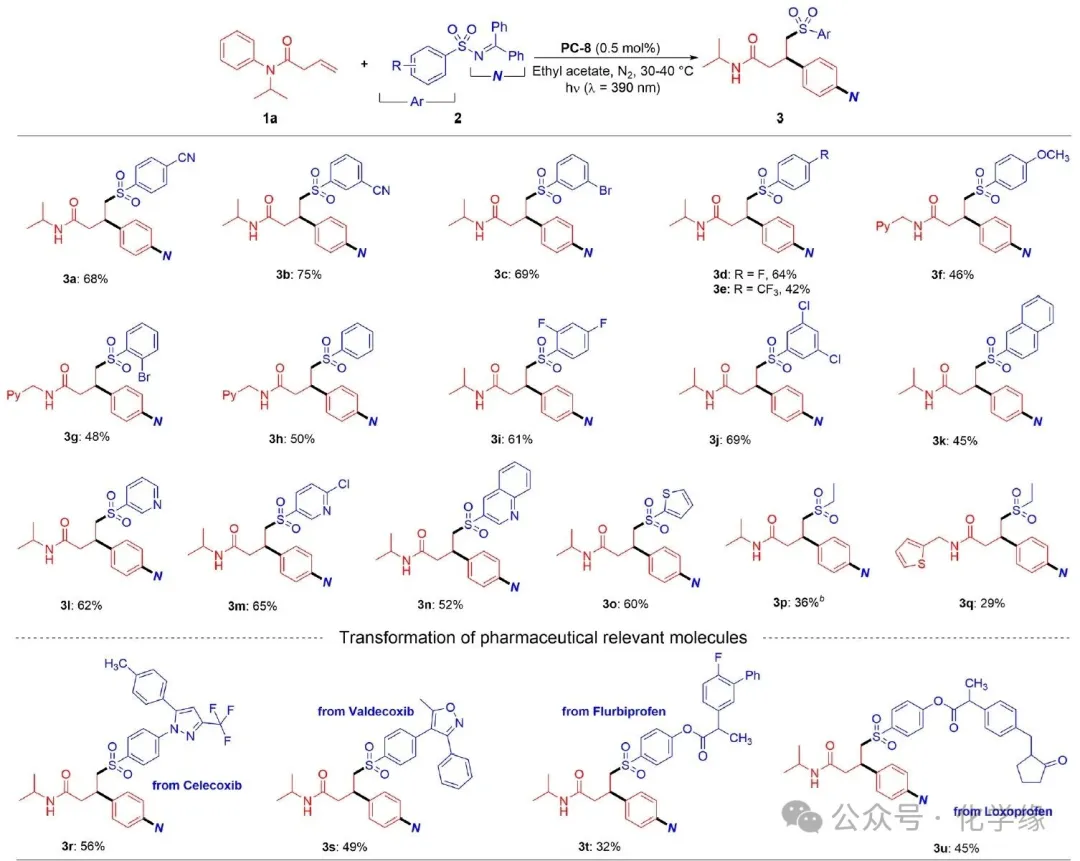
图 3 双功能试剂磺酰亚胺的底物范围
Reaction conditions: 1 (0.1 mmol), 2(0.15 mmol), PC-8 (0.5 mol %), and EtOAc (2 mL) were irradiated in 390 nm LED light for 10 h under N2 atmosphere, 30∼40 °C.
在确定最佳反应条件后,研究当前光催化胺化/迁移转化反应的普适性。首先以酰胺1a作为模型反应物,考察了磺酰亚胺2的反应范围。这些反应对磺酰亚胺展现出广泛的官能团耐受性,不受电子效应影响。例如,当磺酰亚胺对位存在吸电子基团(如氰基、氟原子和三氟甲基)时,反应仍能顺利进行,以中等至良好的收率得到相应的胺化苯丙酰胺。间位氰基和溴原子同样具有良好耐受性,分别以75%和69%的收率得到对应产物。对位强供电子的甲氧基仅导致收率轻微下降,以46%收率得到产物。
磺酰基的立体效应对反应结果影响甚微。例如,磺酰基中心邻位含溴的磺酰亚胺反应顺利进行,产物收率略有降低。二卤代磺酰亚胺(如2,4-二氟磺酰亚胺和3,5-二氯磺酰亚胺)成功重排为相应的苯丙酰胺,收率分别为61%和69%。这些分子中的卤原子为后续官能团化反应提供了可能性。杂环结构在医药和农用化学品中普遍存在,因此将其引入有机分子备受关注。含杂环的磺酰亚胺(包括吡啶环、喹啉环和噻吩环结构)表现出良好的耐受性,对应的含杂环目标化合物分别以52-65%的收率获得。脂肪族磺酰基自由基通常会经历快速脱硫过程生成烷基自由基物种,这为脂肪族磺酰基自由基向烯烃的自由基加成反应带来了重大挑战。尽管如此,乙烷磺酰亚胺仍是当前转化中有效的双功能试剂,但其收率显著降低。
为验证该策略的实用性,采用标准反应条件处理了若干药物相关分子。含磺酰胺基团的药物Celecoxib和Valdecoxib被转化为磺酰亚胺,经本反应后分别以56%和49%的收率得到对应产物。当前反应实现了磺酰胺通过脱氨基连接至高价值的4-氨基化苯丙酰胺,为相关杂合药物提供了快速合成途径。Flurbiprofen和Loxoprofen衍生的磺酰亚胺同样是有效的底物,尽管收率略低。
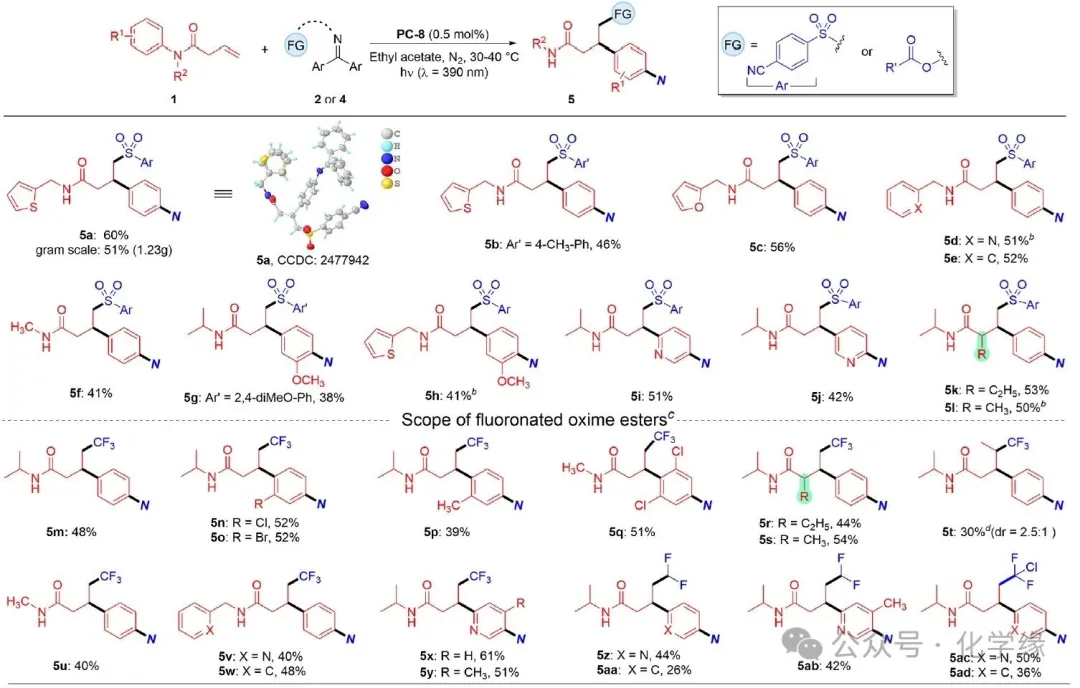
图 4 酰胺与肟酯的底物范围
Reaction conditions: 1 (0.1 mmol), 2(0.15 mmol), PC-8 (0.5 mol %), and EtOAc (2 mL) were irradiated in 390 nm LED light for 10 h under N2 atmosphere, 30∼40 °C.
以磺酰亚胺2a作为模型反应物,探讨了该反应对β,γ-不饱和羧酰胺1的普适性。最初认为大位阻的异丙基基团有助于酰胺1a在自由基环化步骤中得到几何上有利的构象。首先研究了其他氮保护基团(R2)的普适性。将异丙基替换为噻吩-2-基甲基后,分别以60%和46%的收率得到对应的产物。为验证反应的实用性,将5a生产规模扩大至克级,其收率与小试相当。其他保护基(如呋喃-2-甲基基团、吡啶-2-甲基、苄基及甲基)均能耐受该反应,对应产物收率处于可接受至中等水平。通过适当增加催化剂用量并延长反应时间,可略微提高收率(51%)。
无论电子效应如何,N-保护苯环上的取代基对当前反应均具有显著影响。在众多测试的邻位或间位取代基中,仅间位带有甲氧基的芳基团能获得可接受的收率。该现象可能归因于取代基对环化反应及后续自由基重组步骤产生的立体位阻和电子效应。该反应成功应用于吡啶环的迁移与官能团化。通过选用不同起始原料可获得取代基位置相反的两个二取代吡啶化合物。β,γ-不饱和酰胺α位取代基对反应影响甚微,含乙基和甲基的产物分别以53%和50%的收率获得。由于氟元素及其烷基衍生物赋予的独特性质,将氟烷基基团引入目标有机分子长期以来备受合成化学、医学及材料科学领域关注。因此尝试将现有策略扩展至烯烃的三氟甲基化/芳基化反应,以制备含氟烷基基团的氨基化苯丙胺。肟酯作为强效双功能试剂崭露头角,可通过三线态能量转移过程实现烯烃的邻位双官能团化。
选取三氟乙酰肟4a作为双功能试剂与酰胺1a反应。该反应顺利进行,以48%的收率得到含三氟甲基的预期产物。当前的烯烃三氟甲基化/芳基化反应对酰胺芳环上取代基的耐受性更佳。例如,含氯/溴原子或邻位甲基的酰胺在螺环碳原子上均能顺利反应,分别以52%、52%和39%的收率得到相应的胺化苯丙胺。2,6-二氯苯胺衍生的酰胺虽在螺环化步骤存在显著立体位阻,仍作为有效底物,以51%收率得到目标产物。α-及γ-取代酰胺均表现出良好反应性,成功获得对应目标物。相对低收率可能归因于甲基在自由基加成或自由基环化步骤中的位阻效应。此外,由其他含氟酸(如二氟乙酸和氯二氟乙酸)衍生的肟酯被证实为有效底物,可用于制备含二氟甲基或氯二氟甲基的苯丙胺酰胺。
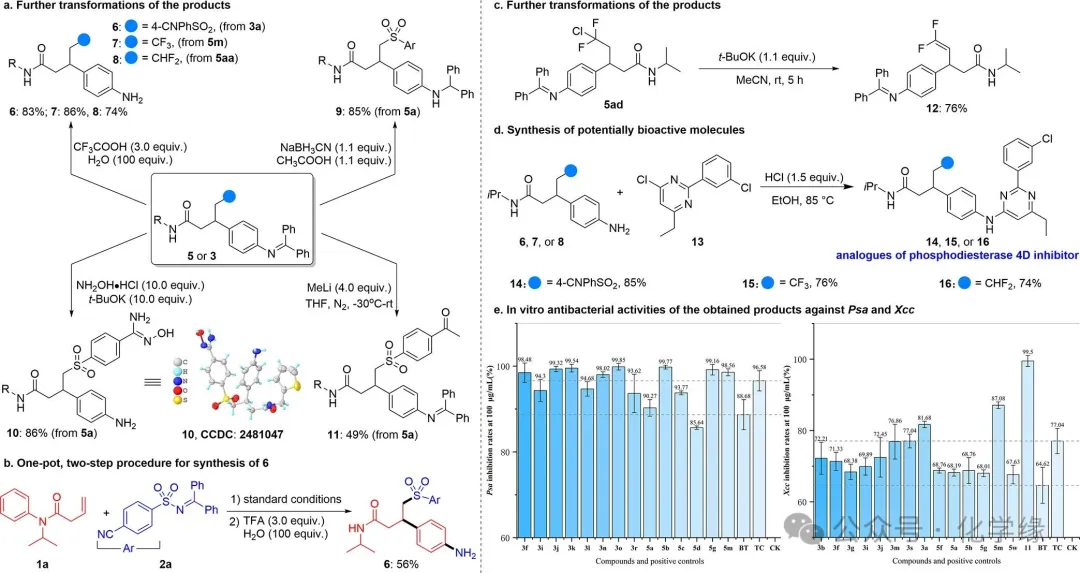
图 5 反应的应用
该产物含有多种官能团,可进行多样化的后续转化。例如,在三氟乙酸存在下,亚胺基团可转化为氨基,分别以83%、86%和74%的收率生成具有磺酰基、三氟甲基或二氟甲基的4-氨基苯基丙酰胺。
为进一步提升方法实用性,以1a为起始原料,采用整合芳基迁移与水解步骤的一锅两步法。目标产物以56%收率获得,与两步操作的收率一致。此外,通过NaBH3CN还原C-N双键可将亚胺基转化为二级胺基团,以85%收率得到9。化合物5a中的氰基可在等当量叔丁醇钾作为碱存在下与盐酸肼反应,以86%收率生成(Z)-N′-羟基苯并咪酰胺10。同时,5a中的亚胺基被转化为氨基。氰基也可与甲基锂等亲核试剂反应,以49%的收率生成酮类产物11。将5ad与叔丁基氧基锂(1.1当量)在室温下于甲基氰化物中反应5小时,经氯化氢消除反应,以76%的收率得到具有价值的对位二氟烯烃12。鉴于3-(4-氨基苯基)丙酰胺基团在生物活性分子中的广泛应用,将其应用于多种磷酸二酯酶4D抑制剂类似物的分支合成。将上述获得的苯胺与预制氯代嘧啶13在1.5当量盐酸存在下反应,分别以85%、76%和74%的收率得到对应产物。
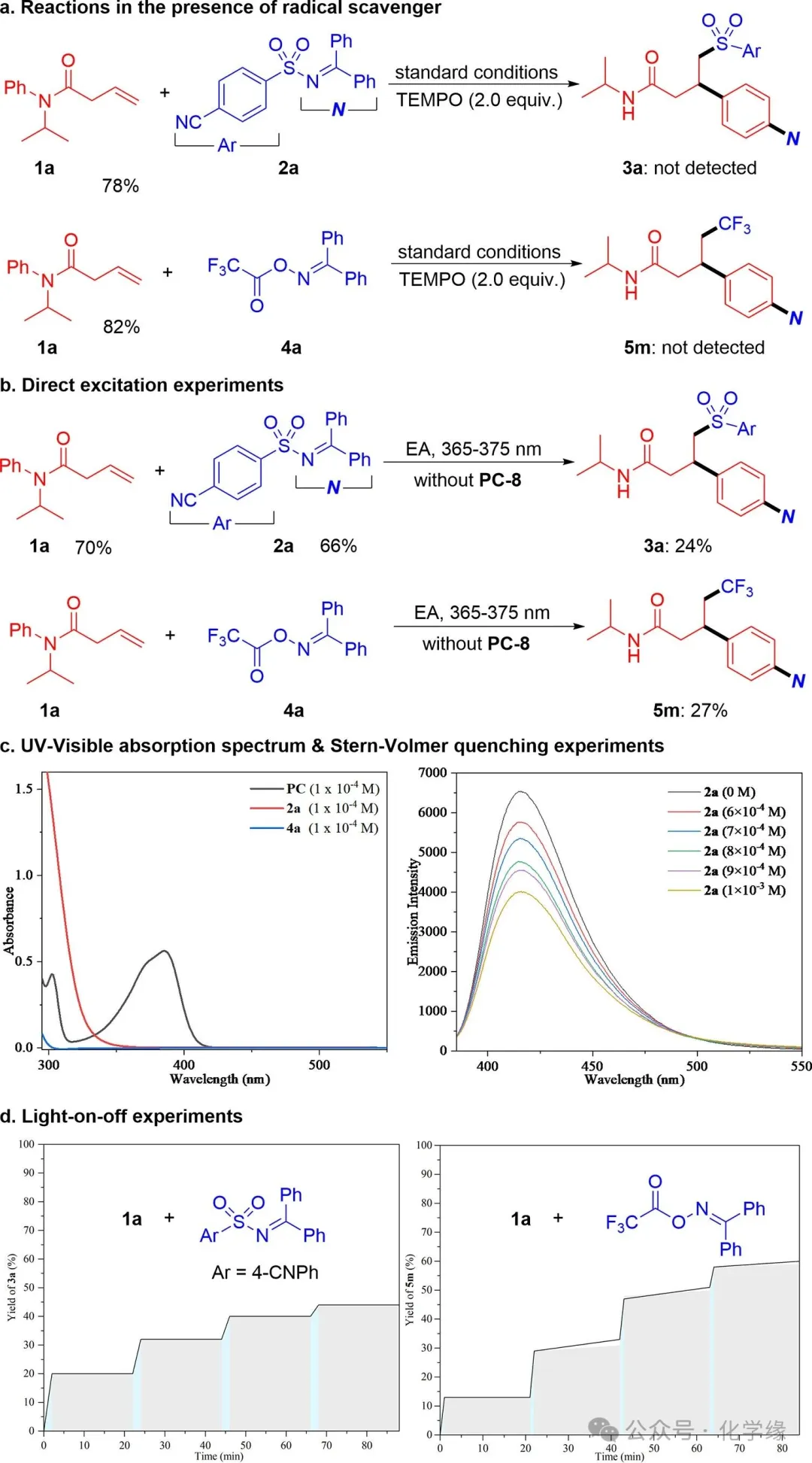
图 6 机理研究
为深入理解反应路径,开展了多项实验研究。首先在完全相同的条件下向模型反应体系中添加TEMPO作为自由基捕获剂。结果显示,芳基磺酰化反应与三氟甲基芳基化反应均被完全抑制,表明当前反应具有自由基性质。通过底物三线态状态进行的反应可通过直接光激发实现,这与单电子转移途径的反应存在本质区别。因此,在不使用光敏剂PC-8的情况下,采用短波长光(λ=365-375 nm)进行了反应。如预期所示,目标产物3a和5m成功获得,但收率显著降低,证实了能量转移途径。紫外-可见吸收光谱显示仅PC-8在390 nm处存在吸收峰。噻吨酮型催化剂与磺酰亚胺及肟酯的三重态能量匹配,有利于PC-8与双功能试剂间的能量转移过程。选取磺酰亚胺2a作为示例,与PC-8共同进行Stern-Volmer淬灭实验,磺酰亚胺2a能有效淬灭PC-8的发光。最终的光开关实验表明,磺酰亚胺与肟酯在黑暗环境中的反应行为存在差异。肟酯反应中略微提高的产率支持其涉及自由基链式反应途径。相反,磺酰亚胺反应即使在黑暗中长时间放置也未发生反应。然而,这不足以排除链式传播机制的可能性,因为自由基链可能衰减过快,无法在实验室时间尺度上检测到。
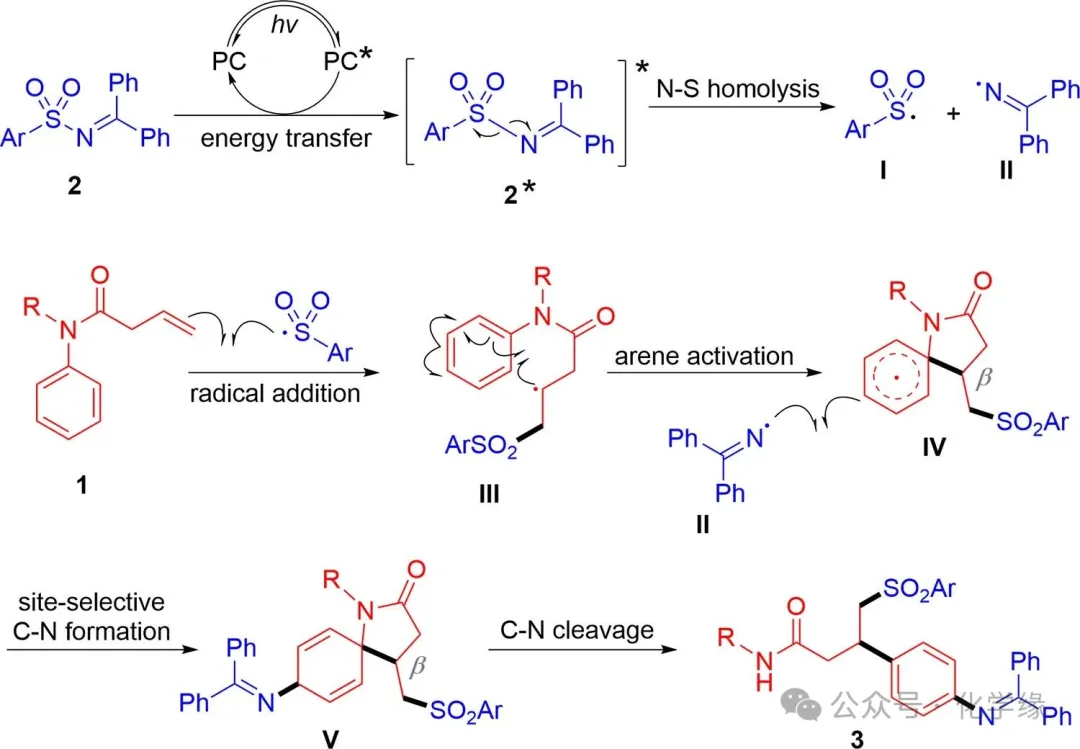
图 7 可能的机理
基于上述实验研究及文献报道,以磺酰亚胺为例,提出并描述了一种可能的反应路径。反应始于光敏剂的光激发,随后与磺酰亚胺2发生能量转移,生成激发态2*,该激发态经N-S键同解键反应生成磺酰自由基I和亚胺自由基II。磺酰自由基I通过自由基加成作用于酰胺1的C-C双键,形成带芳环的碳自由基III。该自由基在芳环内发生异位加成,实现芳环对位超远端活化,生成螺环状中间体IV。随后通过位点选择性自由基重组,在芳环对位形成新的C-N键,得到螺环状环己二烯中间体V。最终,中间体V通过断裂原有的C-N键,在无能量障碍的情况下经历再芳构化步骤,生成最终产物4-氨基苯丙胺3。
总结:开发了一种基于光催化芳基迁移的烯烃双官能团化方法,可快速获得具有价值的4-氨基苯丙酰胺骨架。该反应通过自由基介导的远程芳烃C-H胺化反应实现4-氨基芳环的原位形成,显著提升了传统芳基迁移策略的灵活性,并提高了4-氨基芳基的迁移效率。该反应适用于多种双功能试剂,可实现含磺酰基、三氟甲基、二氟甲基或氯二氟甲基的苯丙酰胺的发散性合成,收率中等至良好。通过对药用相关分子的后期修饰、产物进一步转化、潜在生物活性分子的合成及抗菌活性研究,验证了该策略的实用价值。该策略将为设计新型芳基迁移合成方法提供新思路,并加速复杂结构的合成进程。
来源于:化学缘
文章信息:
Photocatalytic In Situ Amination of the Migrating Aryl Group: Rapid Access to 4-Aminated Benzenepropanamides
Dongmei Chen, Ting Tu, ianhui Liao, Donghan Liu, Shi-Chao Ren*, Yonggui Robin Chi*
DOI: 10.1021/acscatal.5c06638