【转载】绿农黔沿‖贵州大学池永贵/任世超团队:光催化迁移芳基原位胺化策略——高效合成4-胺基苯丙酰胺衍生物
2025-12-22 浏览次数: 54
绿农黔沿‖贵州大学池永贵/任世超团队:光催化迁移芳基原位胺化策略——高效合成4-胺基苯丙酰胺衍生物
4-胺基苯丙酰胺骨架是一类具有重要生物活性的优势药效团,广泛存在于抗肿瘤、抗白血病、抗登革热及西尼罗河病毒等药物分子中,因此开发温和、高效的合成方法构建该类骨架具有重要的应用价值。芳基迁移介导的烯烃双官能化反应是快速构建苯丙酰胺骨架的直接策略之一,该类反应通常由预安装芳基的烯烃与原位生成的自由基物种反应,经芳基迁移过程实现芳基的引入。然而,传统策略存在显著局限:一方面,特定芳基的预安装步骤往往合成复杂,限制了方法的通用性;另一方面,迁移过程中芳环的原位官能化一直难以实现,主要原因是传统金属催化的芳环 C−H官能化与自由基介导的芳基迁移过程存在兼容性问题,无法在同一反应体系中协同进行,极大限制了芳基迁移策略的灵活性和迁移效率。
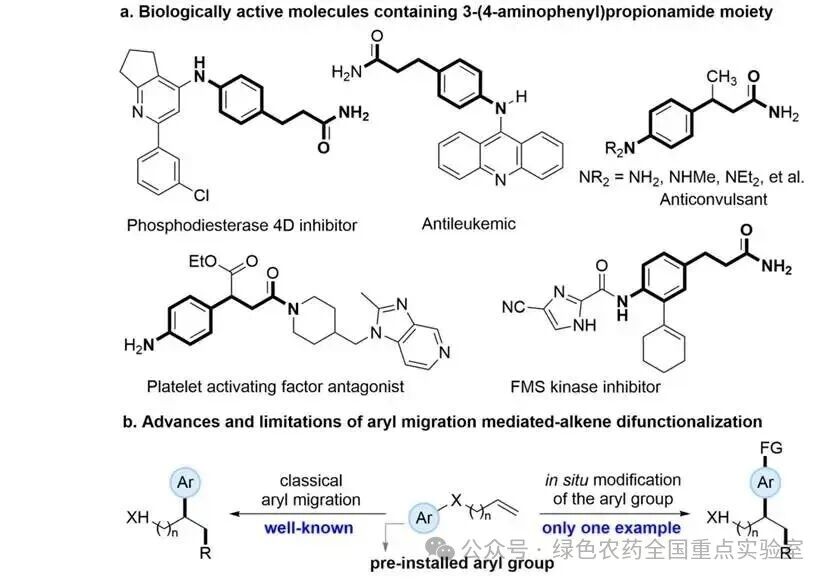
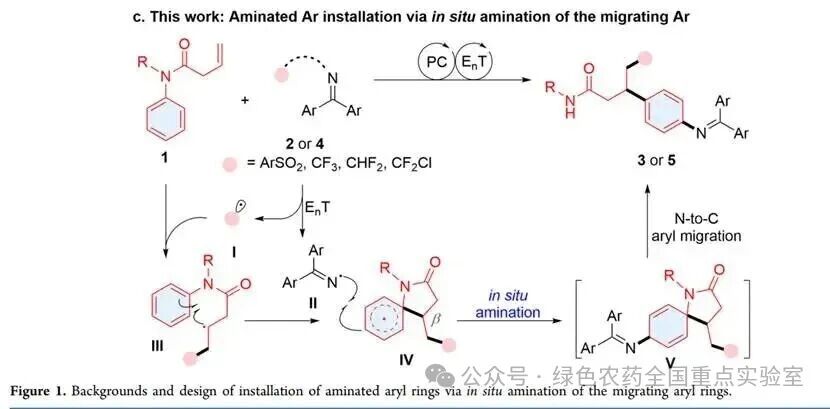
本文通过光催化实现迁移芳环的原位胺化,将芳环胺化与迁移过程整合于单步反应中,成功突破了传统方法的核心局限。其核心创新在于利用光敏剂与双功能试剂之间的能量转移催化循环,将芳基迁移过程与新兴的自由基介导的芳环远程 C−H胺化步骤相结合,在烯烃碳碳双键形成两根新键的基础上,进一步在迁移芳环上构建额外的 C−N键,显著提升了芳基迁移策略的灵活性和胺化芳环的迁移效率。该策略展现出三大核心优势:1. 一步构建多根化学键:同步实现芳基迁移与芳环原位胺化,直接得到 4 - 胺基苯丙酰胺骨架,无需冗长的预官能化步骤;2. 底物适用范围广泛:兼容磺酰亚胺、氟代肟酯等多种双功能试剂,可合成含磺酰基、三氟甲基、二氟甲基等不同官能团的目标产物;3. 实用性强:可应用于药物相关分子的后期修饰、生物活性分子类似物的发散合成,且产物具有良好的抗菌活性。
研究团队首先针对反应的核心挑战展开探索:需筛选合适的双功能试剂,使其能同时生成两种反应性不同的自由基(瞬态自由基参与烯烃加成,持续亚胺自由基主导原位胺化);同时需解决芳基迁移与芳环C−H胺化的兼容性问题,确保两步反应在同一体系中高效协同进行。
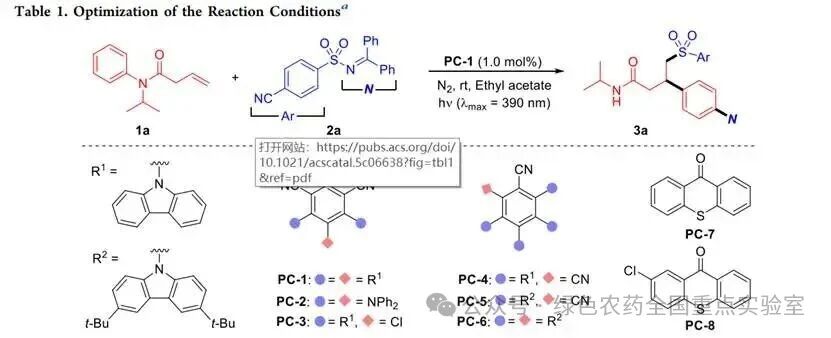
团队以C=C双键连接的 N - 苯基酰胺1a为模板底物,磺酰亚胺2a为双官能试剂,对反应条件进行了系统优化。初始尝试以4CzIPN 为光敏剂、乙酸乙酯为溶剂,在 390 nm LED 光照下反应 6 小时,目标产物 3a 收率仅 20%。通过筛选多种氰基苯衍生光敏剂发现,五咔唑氰基苯PC-6催化效率显著提升,收率达54%;进一步尝试硫杂蒽酮类光敏剂(PC-7、PC-8),其中2-氯硫杂蒽酮 PC-8 表现最佳,收率提升至64%。溶剂筛选显示,乙酸乙酯、乙酸甲酯和二氯甲烷均适用,而四氢呋喃、乙腈和二甲基甲酰胺会导致收率显著下降。降低光敏剂负载至0.5 mol%,收率保持不变;将反应时间延长至10小时,收率进一步提升至71%,分离收率达68%。对照实验证实,光敏剂是反应必需的,无光敏剂存在时反应完全无法进行。
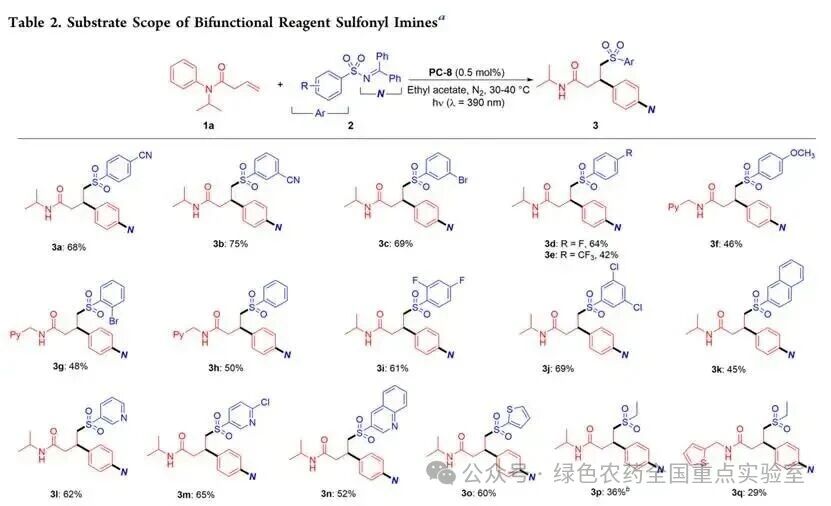
在优化条件基础上,团队首先拓展了双官能试剂磺酰亚胺的适用范围。无论磺酰亚胺芳环上取代基的电子性质如何,反应均能顺利进行:对位含氰基、氟原子、三氟甲基等吸电子基团的磺酰亚胺反应收率良好(3a、3d、3e);间位含氰基、溴原子的磺酰亚胺收率分别为 75% 和 69%(3b、3c);对位含强给电子甲氧基的磺酰亚胺收率略有下降,为 46%(3f)。空间位阻对反应影响较小,磺酰基相邻位置含溴原子的底物仍能顺利反应(3g);二卤代磺酰亚胺(2,4 - 二氟、3,5 - 二氯)均能高效转化,收率 61%-69%(3i、3j),产物中的卤素原子为后续官能化提供了可能。杂芳环磺酰亚胺同样适用,含吡啶、喹啉、噻吩环的底物收率 52%-65%(3l-3o)。脂肪族乙磺酰亚胺虽能参与反应,但收率显著降低(3p、3q)。
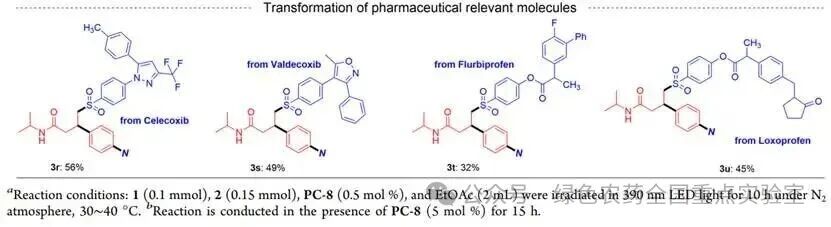
为验证方法的实用性,团队将药物相关分子衍生的磺酰亚胺应用于反应:塞来昔布、伐地昔布衍生的磺酰亚胺反应收率分别为56%和49%(3r、3s);氟比洛芬、洛索洛芬衍生的磺酰亚胺也能顺利反应,收率32%-45%(3t、3u),实现了药物分子的后期修饰与复杂药物分子的快速构建。
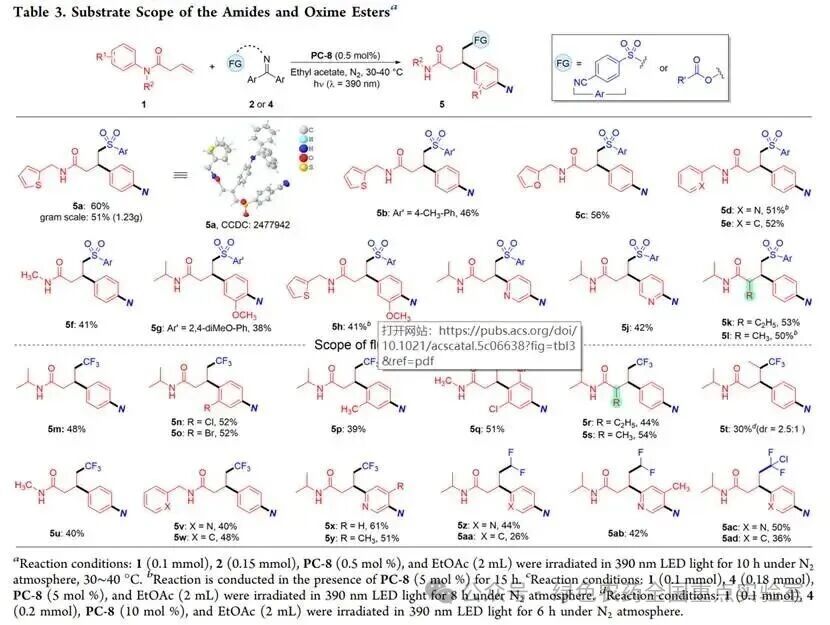
随后,团队拓展了酰胺底物与肟酯类双功能试剂的适用范围。对于 β,γ- 不饱和羧酸酰胺 1,不同氮保护基均能兼容,收率 41%-60%(5a-5f);其中产物5a的结构经X射线单晶衍射确认(CCDC: 2477942),且该反应可放大至克级规模,收率保持51%(1.23 g)。N - 保护苯环上的取代基对反应有一定影响,仅间位含甲氧基的底物能获得可接受收率(5g、5h);吡啶环作为迁移芳环时,通过选择不同起始原料可得到两种取代基位置相反的二取代吡啶产物(5i、5j);酰胺 α 位含乙基、甲基的底物反应收率稳定(5k、5l)。
针对含氟烷基官能团的目标产物合成,团队选用三氟乙酰肟酯4a等氟代肟酯作为双官能试剂,成功实现烯烃的三氟甲基化/芳基化反应,收率48%(5m)。该体系对酰胺芳环上的取代基耐受性良好,含氯、溴、甲基的底物,甚至2,6 -二氯苯胺衍生的位阻较大的酰胺,均能高效反应(5n-5q);α- 和 γ- 取代酰胺也能顺利转化(5r-5t)。此外,二氟乙酸、氯二氟乙酸衍生的肟酯可用于合成含二氟甲基、氯二氟甲基的苯丙酰胺衍生物(5z-5ad),进一步丰富了产物的结构多样性。
该方法合成的产物含多个官能团,可进行多样化后续转化:亚胺基团在三氟乙酸作用下可转化为氨基,收率 74%-86%(6-8);通过一锅两步法整合芳基迁移与水解步骤,产物 6 收率达 56%,与分步反应相当;亚胺基团经 NaBH₃CN 还原可生成仲胺(9,收率 85%);产物 5a 中的氰基可与盐酸羟胺反应生成(Z)-N'- 羟基苯甲脒(10,收率 86%),或与甲基锂反应生成酮类产物(11,收率 49%);化合物 5ad 经叔丁醇钾处理可消除氯化氢,生成含偕二氟烯烃结构的产物(12,收率 76%)。将产物进一步应用于生物活性分子类似物的合成:所得苯胺类化合物 6-8 与预制备的氯化嘧啶13反应,成功合成含磺酰基、三氟甲基、二氟甲基的磷酸二酯酶4D抑制剂类似物,收率74%-85%(14-16)。抗菌活性测试显示,多数产物在 100 μg/mL 浓度下对野油菜黄单胞菌和猕猴桃溃疡病菌表现出优异的抑制活性,优于商用杀菌剂噻菌铜和氢氧化铜,具有开发为新型农作物保护杀菌剂的潜力。
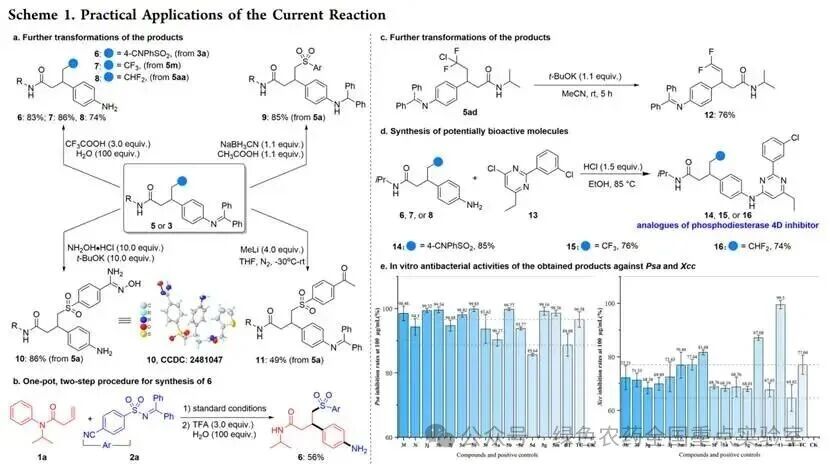
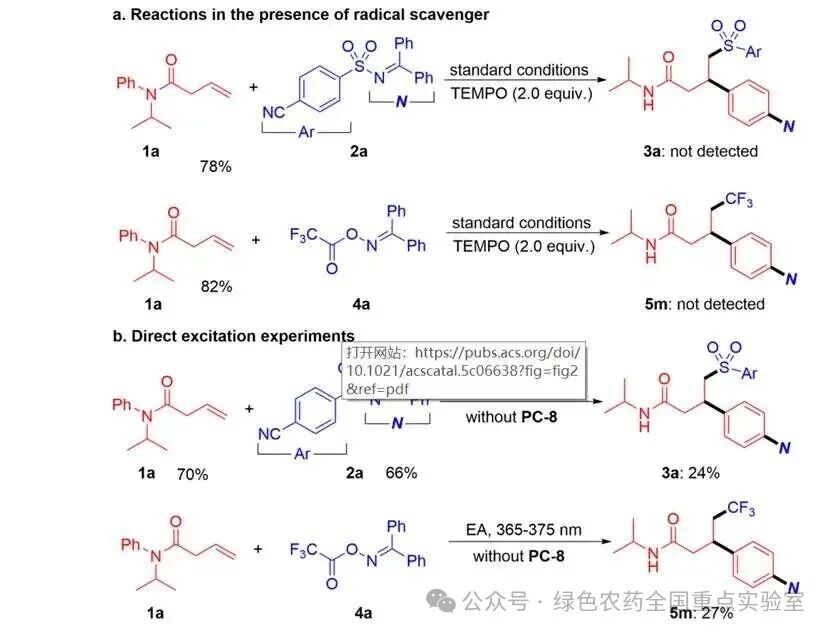
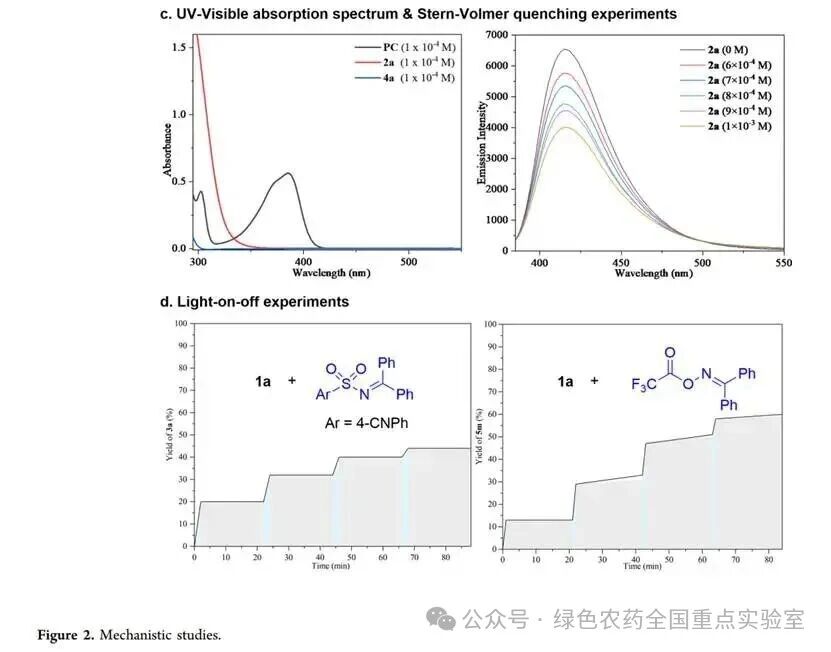
团队通过一系列机理实验对反应路径进行了验证:向模型反应中加入自由基捕获剂 TEMPO,芳基磺酰化和三氟甲基化 / 芳基化反应均被完全抑制,证实反应具有自由基特性;在无光敏剂存在下,使用 365-375 nm 短波光照仍能得到目标产物(3a、5m),但收率显著下降,表明反应通过能量转移路径进行;紫外 - 可见吸收光谱显示,仅光敏剂 PC-8 在 390 nm 处有吸收,且其三重态能量与磺酰亚胺、肟酯匹配,有利于能量转移;Stern-Volmer 猝灭实验证实,PC-8 的发射可被磺酰亚胺 2a 有效猝灭;光开关实验表明,磺酰亚胺参与的反应在黑暗中无进展,而肟酯参与的反应在黑暗中收率略有提升,提示后者可能涉及自由基链路径。
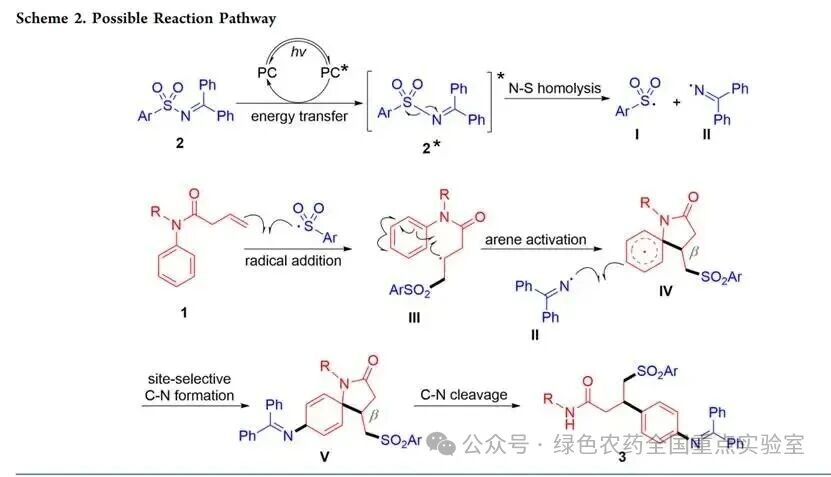
基于上述实验结果和相关文献,团队提出了可能的反应机理:首先,光敏剂经光照激发,通过能量转移使磺酰亚胺2处于激发态(2*),其 N−S 键均裂生成磺酰自由基I和亚胺自由基II;磺酰自由基I与酰胺1的C=C双键发生自由基加成,形成芳环连接的碳自由基III;该自由基与芳环发生分子内自由基同位加成,激活芳环对位,生成螺环中间体IV;随后发生位点选择性自由基重组,在芳环对位形成新的C−N键,得到螺环己二烯中间体V;最后,中间体V通过断裂原有C−N键,经无能量壁垒的再芳构化步骤,生成最终的4 - 胺基苯丙酰胺产物 3,并完成催化循环。(来源:岳麓化学)