Angew Chem:贵州大学伍星星教授等突破 S (VI) 手性农药合成瓶颈:异硫脲催化新策略,高效构建抗菌活性磺酰亚胺衍生物
2025-09-26 浏览次数: 13
在有机合成领域,“手性” 是决定分子功能的关键密码——从药物的疗效到材料的性能,手性分子的精准构建一直是研究者追逐的核心目标。其中,含硫手性化合物因其在药物化学、农业化学中的广泛应用(比如作为抗菌、抗炎药效团),更是备受关注。
近年来,硫 (IV)(S (IV))手性化合物(如亚砜、亚磺酰胺)的合成已取得长足进展,Ellman、Willis等名家团队的工作让这一领域日趋成熟。但与之形成鲜明对比的是,硫 (VI)(S (VI))手性化合物——这类具有sp³杂化、构型稳定的四取代硫中心分子,却长期处于 “研究洼地”。
为何 S (VI)手性如此难啃?核心痛点有三:一是S (VI)中心构型异常稳定,难以实现催化控制下的构型翻转;二是现有方法多依赖手性S (IV)前体的氧化,底物多样性受限;三是直接从外消旋 S (VI)原料出发的催化不对称合成,始终缺乏高效策略。
近期,贵州大学伍星星教授、池永贵教授团队在国际顶刊《Angewandte Chemie International Edition》(Angew)以 “Hot Paper”形式发表了一项突破性研究,不仅解决了 S (VI)手性催化的核心难题,还为抗菌农药的开发提供了新方向。
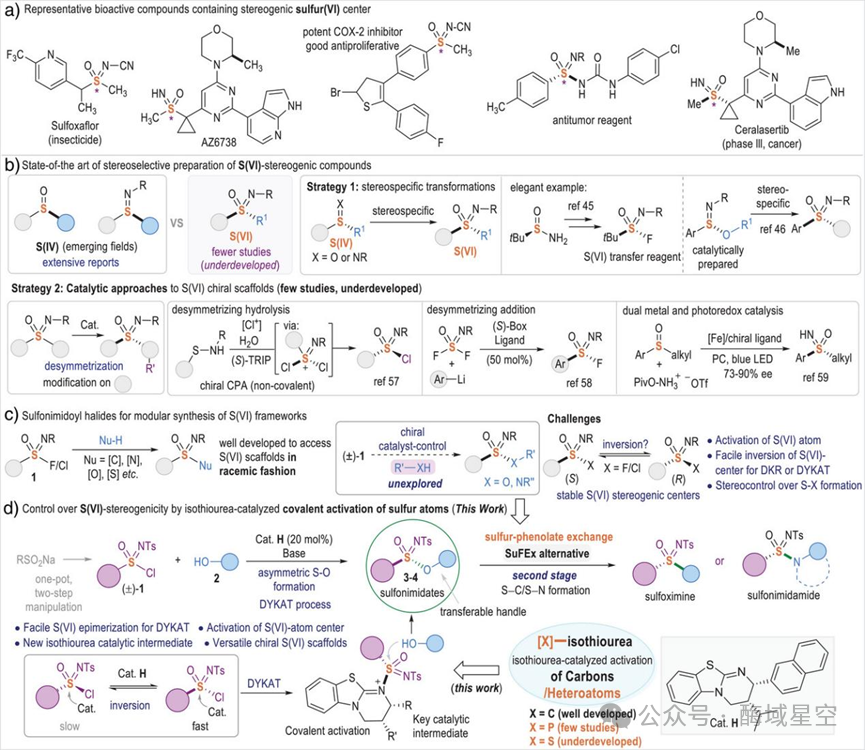
图1:从亚磺酸衍生物合成手性亚硫酸盐的催化剂控制策略
实验结果
一、核心突破:异硫脲催化的 “跨界操作”—— 从C中心到S中心的活化革命
有机催化中,异硫脲是一类 “明星催化剂”——它擅长通过共价活化羧酸、酯等碳中心(C-中心)底物,形成酰基铵等活性中间体,进而实现高选择性反应。但在此之前,异硫脲催化从未被成功应用于硫中心(S-中心) 的活化。
而这项研究的第一个 “神操作”,就是将异硫脲的催化能力 “跨界” 到 S (VI)原子上:通过与外消旋磺酰亚胺氯(S (VI)原料)形成异硫脲结合的磺酰亚胺中间体,实现了S (VI)原子的共价活化。
这个中间体有多关键?它不仅降低了 S (VI)中心的反应能垒,还为后续的不对称反应搭建了 “手性模板”—— 当酚类亲核试剂进攻时,中间体的手性环境能精准控制 S-O键的形成,最终以高达 97:3的对映选择性和 86%的产率得到 S (VI)手性磺酰亚胺酯。
更重要的是,作者还引入了动态动力学不对称转化(DYKAT) 机制:在异硫脲催化剂的辅助下,原本稳定的 S (VI)手性中心能实现 “ facile inversion(易翻转)”—— 外消旋原料中的两种构型(R/S)可以快速互变,最终全部转化为热力学更稳定的单一构型产物。这一机制彻底打破了传统动力学拆分 “产率不超过 50%”的局限,实现了 “产率 +选择性” 双丰收。
二、实力验证:底物范围 + 转化能力,尽显 “合成平台” 价值
一项优秀的合成方法,不仅要 “巧”,更要 “实用”。吴兴兴团队通过大量实验,充分验证了该策略的普适性和转化潜力,主要体现在两个方面:
1.底物兼容性:从简单分子到药物片段,无一不 “通”
作者首先考察了磺酰亚胺氯的底物范围:无论是芳环上带有甲基、乙基、叔丁基等烷基,还是氟、溴、碘等卤素,甚至是三氟甲基、三氟甲氧基等强吸电子基团,反应都能顺利进行,得到对应的磺酰亚胺酯(3a-3j),对映选择性稳定在 91:9~97:3之间。
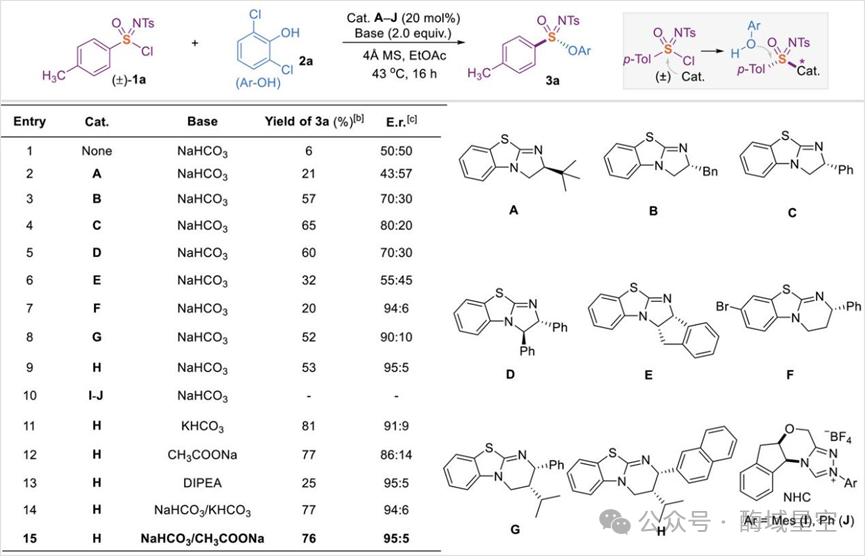
图2:磺酰亚胺酯 3a的对映选择性合成优化
更值得关注的是空间位阻的耐受性:当芳环邻位有取代基(如 3k-3m),甚至是位阻极大的均三甲苯基(3q)时,反应依然能保持高选择性 —— 这意味着该方法可以用于构建结构复杂的 S (VI)手性分子。
对于酚类亲核试剂,兼容性同样出色:从简单的苯酚(4a),到带有硝基、多卤素取代的 “电子缺” 酚(4b-4e),再到五取代的 “拥挤” 酚(4j),均能高效反应。
最亮眼的是天然产物和药物分子的后期修饰:作者成功将该方法应用于 7 -羟基香豆素(4k,抗凝血药物前体)、对乙酰氨基酚(4m,退烧药主要成分)、乙炔雌二醇(4p,激素类药物)、紫檀芪(4q,抗氧化剂)等分子的磺酰亚胺化修饰,产率高达 67%~90%,对映选择性几乎无损失。这为药物分子的手性衍生化提供了全新工具。
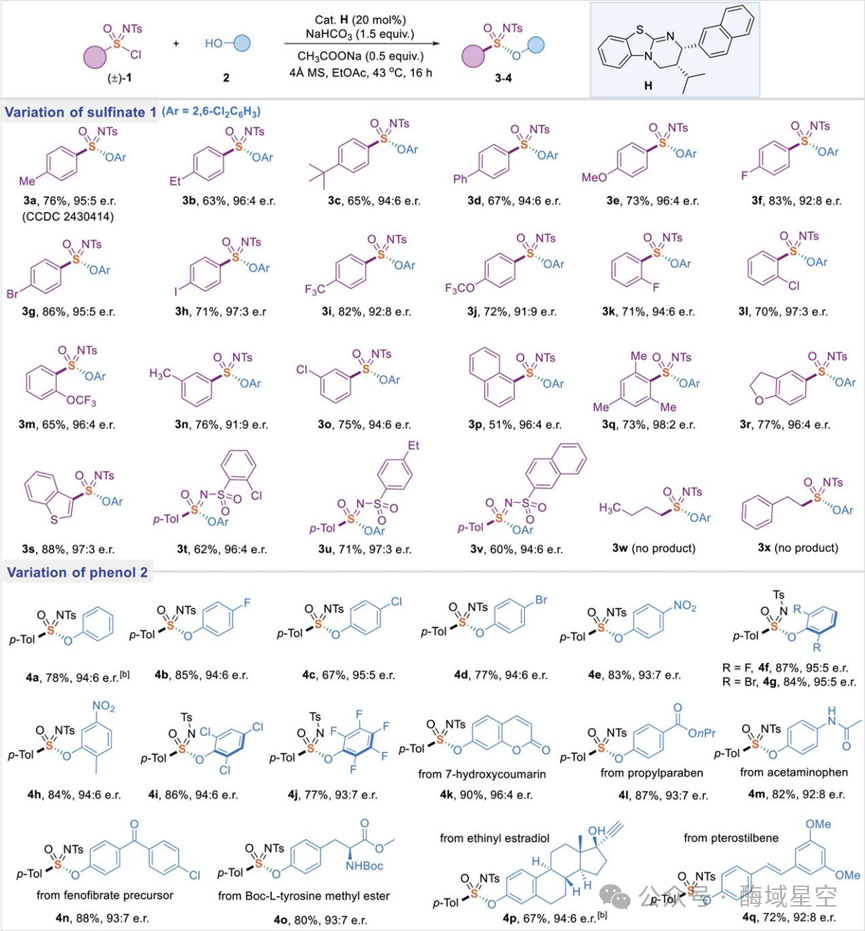
图3:磺酰亚胺酯 3-4的底物范围
2.转化能力:“一键” 构建多种 S (VI)手性骨架,替代 SuFEx
合成出磺酰亚胺酯只是第一步 —— 作者发现,这类化合物是名副其实的 “万能中间体”:通过硫 -酚盐交换反应(作为 SuFEx的温和替代方案),可以快速转化为其他高价值的 S (VI)手性化合物。
合成磺肟(6系列):向磺酰亚胺酯中加入格氏试剂、有机锂试剂等亲核试剂,能以高立体专一性得到磺肟(6a-6k)。无论是烷基、芳基,还是含双键、杂环的亲核试剂,都能顺利反应,产率 73%~94%,对映选择性保持在 95:5以上。
合成磺酰亚胺酰胺(8系列):用胺类亲核试剂取代酚氧基,可得到磺酰亚胺酰胺。作者甚至将该反应应用于天然生物碱(金雀花碱,8l)、商用药物(苯佐卡因,8m;地氯雷他定,8n)的修饰,成功引入 S (VI)手性中心 —— 这为新型手性药物的研发打开了大门。
构型翻转与脱保护:通过简单的酚盐交换,还能实现 S (VI)中心的构型翻转(得到 ent-3a);而 N-Ts保护基的脱除,则能得到含 N-H键的磺肟(9),为后续功能化提供 “抓手”。
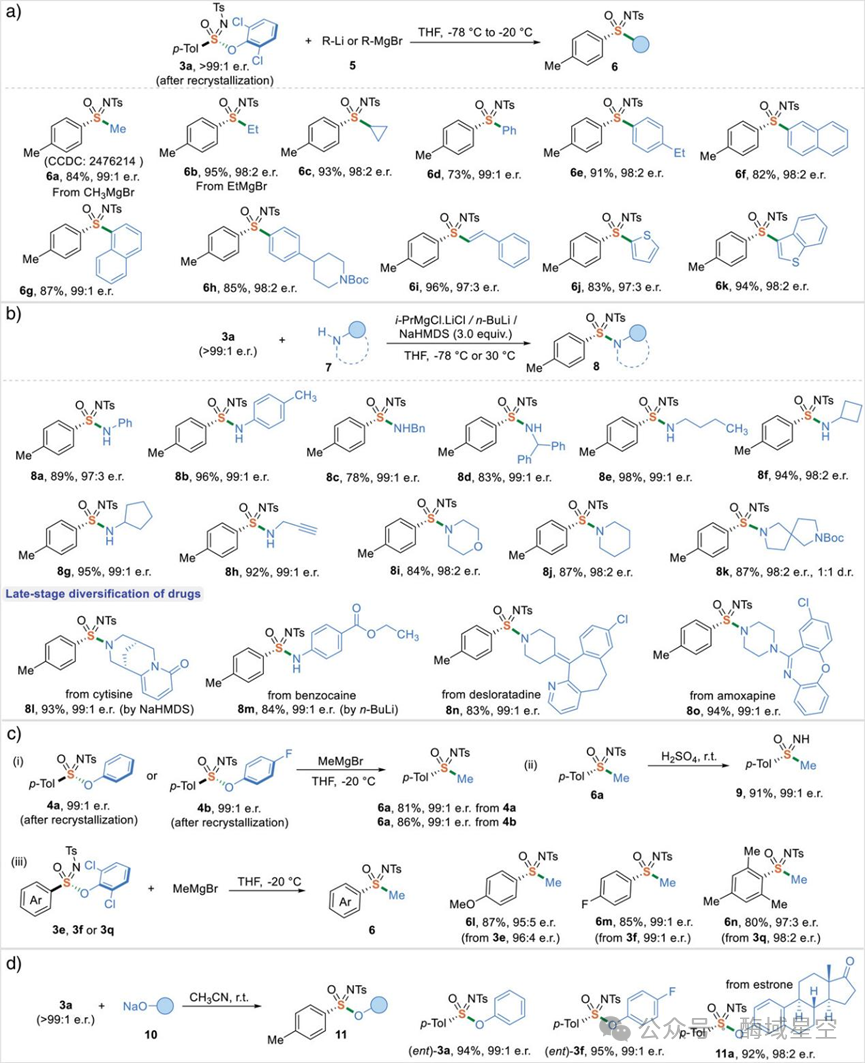
图4:磺酰亚胺酯的硫 (VI)-酚盐交换反应
三、实战检验:抗菌活性亮眼,助力绿色农药开发
一项基础研究的终极价值,在于解决实际问题。吴兴兴团队并未止步于合成,而是进一步考察了这些 S (VI)手性化合物的农业抗菌活性—— 毕竟,植物病原菌(如细菌、真菌)是导致作物减产的主要原因,开发高效、低毒的新型农药迫在眉睫。作者选取了多种常见植物病原菌(如水稻白叶枯病菌 Xoc、番茄灰霉病菌 Bc、小麦纹枯病菌 Rs等),测试了化合物的抑制活性。结果令人惊喜:抗细菌活性:化合物6i对Xoc的抑制率高达70.8%,显著优于商用杀菌剂 TC(62.0%);抗真菌活性:化合物4a对Rs的抑制率达 80.2%,与商用杀菌剂嘧菌酯(azoxystrobin)相当;手性差异:研究还发现,手性构型对活性有显著影响——单一构型的化合物活性普遍优于外消旋体,这也凸显了“手性控制”在农药研发中的重要性。这些结果表明,该方法合成的S (VI)手性化合物,有望成为新型绿色农药的 “候选分子”,为农业病害防治提供新策略。
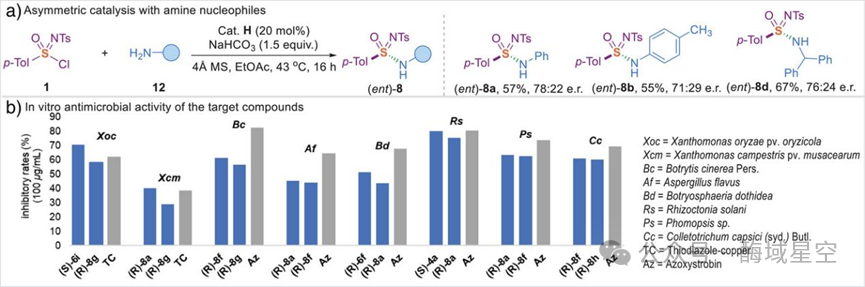
图5: a)磺酰亚胺与胺亲核试剂的催化合成;b)生物测定研究
四、机理揭秘:DFT + 实验,搞懂 “为什么行”
优秀的合成方法,背后必有清晰的机理支撑。为了搞清楚异硫脲催化 S (VI)不对称合成的 “底层逻辑”,作者结合控制实验和密度泛函理论(DFT)计算,还原了反应的完整路径:
中间体形成:外消旋磺酰亚胺氯(1a)与异硫脲催化剂(Cat. H)、碳酸氢钠结合,形成复合物(INT1);催化剂对 S (VI)中心发起亲核进攻,氯离子离去,生成关键的异硫脲-磺酰亚胺中间体(INT2)—这一步得到了高分辨质谱(HRMS)的直接证实。
构型翻转:在催化剂辅助下,INT2的 S (VI)中心可以发生构型翻转(能量垒仅 19.8 kcal/mol),实现 R/S构型的快速互变,为DYKAT机制提供了可能。
选择性决定步骤:酚类亲核试剂进攻 INT2的S (VI)中心,形成过渡态(TS2)。DFT计算显示,生成 (S)-3a的过渡态(S-TS2)能量比生成 (R)-3a的过渡态(R-TS2)低 2.8 kcal/mol——这一能量差正是高对映选择性的来源。
进一步分析过渡态结构发现,S-TS2中存在双重π-π堆叠作用(催化剂与底物之间)和Na-O协同作用(钠离子同时结合底物与碳酸氢根),而R-TS2仅存在单一π-π作用。更强的非共价相互作用让S-TS2更稳定,最终导向(S)-构型产物——这也完美解释了实验结果的选择性。
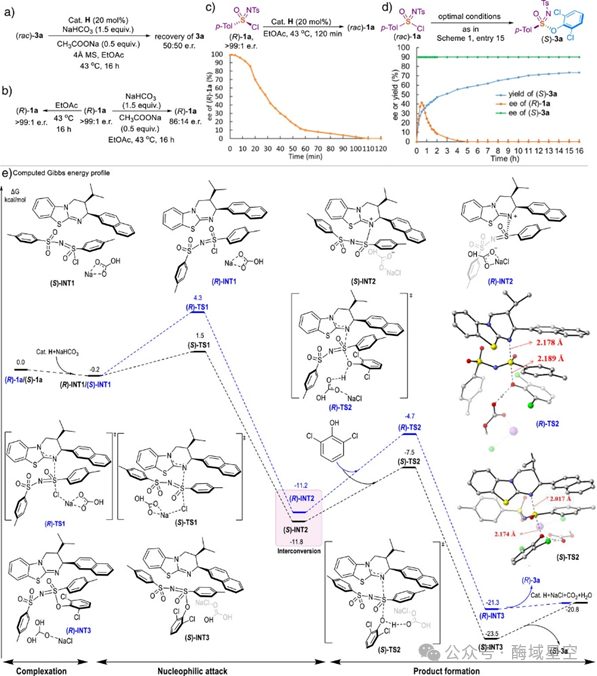
图6:初步机理研究
五、总结:不止于 “合成”,更是未来的 “钥匙”
这篇 Angew Hot Paper之所以精彩,不仅在于它解决了S (VI)手性催化的长期难题,更在于它为有机合成和应用化学提供了多重启示:
催化理念的突破:将异硫脲催化从“C中心”拓展到“S中心”,为杂原子手性催化开辟了新方向——未来,这种“跨界活化”思路或许还能应用于P、N等其他杂原子。
合成平台的价值:通过“一步催化 +多步转化”,实现了多种S (VI)手性骨架的高效构建,为药物、农药研发提供了“模块化”工具——研究者无需重复开发新方法,只需基于磺酰亚胺酯中间体进行衍生。
基础研究到应用的闭环:从解决 “科学难题”(S (VI)手性控制)到落地 “实际需求”(抗菌农药),为年轻研究者展示了基础化学的应用潜力——好的合成方法,不仅要“好看”(高选择性、高产率),更要“好用”(解决产业痛点)。
对于从事有机合成、药物化学、农业化学的研究者来说,这篇论文不仅是一个“新反应”,更是一个“新起点”——它提示我们:那些长期被忽视的“研究洼地”(如 S (VI)手性),或许正是下一个创新突破口。
原文链接:
Li B, Hu J, Xu Z, et al. Control Over S (VI) Stereogenicity for the Asymmetric Synthesis of Sulfonimidoyl Derivatives by Isothiourea-Catalyzed Covalent Activation of Sulfur (VI) Atoms[J]. Angewandte Chemie, 2025: e202510595. DOI:10.1002/anie.202510595
https://onlinelibrary.wiley.com/doi/abs/10.1002/ange.202510595